
A new data analysis approach identifies disease-associated splicing variants
August 24, 2022
Researchers from Tokyo Medical and Dental University (TMDU) find that integrating and selectively analyzing splicing variant data can help identify genetic variations associated with complex human disease
Tokyo, Japan - In the era of Big Data, obtaining a huge amount of information is the easy part; knowing what to do with it is another story entirely. But now, researchers from Japan have reported that a new approach to analyzing data from genome-wide association studies could help uncover the genetic basis of many diseases.
In a study published in August in Nature Communications, researchers from Tokyo Medical and Dental University (TMDU) have revealed that analyzing the coding sequences of gene splicing variants at sites associated with disease can help reveal the genetic cause of certain complex human diseases.
Variations in our genes cause complex diseases, but it can be difficult to tell how a single genetic variation leads to disease. While some variants cause disease by changing gene expression levels, it is increasingly apparent that splicing variants that affect how a gene is transcribed – meaning, how a gene’s DNA sequence is copied into RNA - also play an important role.
“There are a number of existing approaches to identify and analyze genetic variants causing splicing changes in disease-associated genes,” explains Kensuke Yamaguchi, lead author on the study. “However, these approaches are limited by incomplete annotation of splicing isoforms and by the use of the same splicing junction by multiple isoforms, which can make them difficult to distinguish from each other.”
To overcome these drawbacks, the researchers developed a set of two analyses that more fully capture the complexity of splicing variations and their relationship to human disease: the first analysis integrates isoforms with the same coding sequence to detect resulting changes in protein structure, and the second analysis examines the effects of isoforms with incomplete annotations but unique coding sequences. The team then determined the complete sequences of these isoforms and validated their expression in cells.
“The results showed that our approach is both robust and effective,” states Yuta Kochi, senior author on the paper. “We successfully identified 29 full-length isoforms with unannotated coding sequences associated with genetic variants that have been linked to diseases such as Parkinson’s disease, ankylosing spondylitis, irritable bowel disease, and neurodegenerative disease.”
Furthermore, they showed that genes with disease-associated splicing variants can be identified by evaluating their effects on the expression of other genes within the genome. For example, a variant leading to alteration in the ratio of two isoforms of the SNRPC gene was identified as being associated with systemic lupus erythematosus.
Taken together, these findings highlight the unappreciated role of protein-altering splicing variants in causing disease. Identifying relevant variants and assessing their function in future research using animal models could help clarify how complex diseases arise.
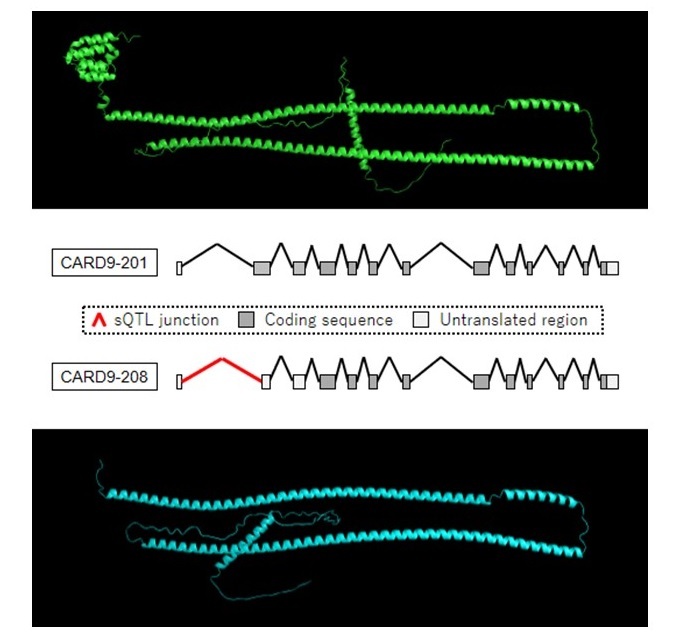
Alternatively spliced isoforms
Alternative splicing of CARD9, regulated by Crohn’s disease risk variant (sQTL variant), changes the protein structure.
###
The article, “Splicing QTL analysis focusing on coding sequences reveals mechanisms for disease susceptibility loci,” was published in Nature Communications at DOI: https://doi.org/10.1038/s41467-022-32358-1
Summary
Researchers from Tokyo Medical and Dental University have found that integrating genetic variants with the same coding sequence and selecting annotated variants with incomplete sequences for further analysis can effectively identify splicing variants that affect protein structure and function. This combined analysis approach has the potential to help explain the genetic basis of complex human disease.
Journal Article
JOURNAL:Nature Communications
TITLE:Splicing QTL analysis focusing on coding sequences reveals the causal mechanisms for disease susceptibility loci
DOI:https://doi.org/10.1038/s41467-022-32358-1
TITLE:Splicing QTL analysis focusing on coding sequences reveals the causal mechanisms for disease susceptibility loci
DOI:https://doi.org/10.1038/s41467-022-32358-1
Correspondence to
Yuta Kochi,MD, Ph.D.,Professor
Department of Genomic Function and Diversity,
Medical Research Institute,
Tokyo Medical and Dental University, (TMDU)
E-mail:y-kochi.gfd(at)mri.tmd.ac.jp
*Please change (at) in e-mail addresses to @ on sending your e-mail to contact personnels.