
"Early Changes to Synapse Gene Regulation May Cause Alzheimer’s Disease"

Hitoshi OKAZAWA, Ph.D.,Professor (Center)
Department of Neuropathology,Medical Research Institute
Kyota FUJITA, Ph.D.,Assistant Professor (Right)
Department of Neuropathology,Medical Research Institute
Hikari TANAKA,Graduate student(Left)
Department of Neuropathology,Medical Research Institute
Tokyo Medical and Dental University (TMDU)-led Japanese researchers identify role for splicing that precedes amyloid plaque formation
Tokyo, Japan – Alzheimer’s disease (AD) is the most common form of dementia, involving memory loss and a reduction in cognitive abilities. Patients with AD develop multiple abnormal protein structures in their brains that are thought to destroy or damage nerve cells (neurons). One of these structures, the senile plaque, is made up of clumps of beta-amyloid (Aβ) peptide which form in the spaces between neurons.
Many advanced clinical trials in patients with AD have attempted to slow down or reverse the disease by targeting these plaques for removal. However, despite the successful decrease in Aβ aggregation, these trials mostly have failed to improve memory or cognitive function in AD patients.
Before the formation of Aβ aggregates, studies revealed changes in the phosphorylation (a chemical modification) of certain proteins, including SRRM2. This protein was thought to be involved in a form of gene regulation known as splicing, but its exact function was unclear.
Now, a Japanese team of researchers led by Tokyo Medical and Dental University (TMDU) has examined levels of SRRM2 phosphorylation in a mouse model of AD, and found that they increased prior to Aβ aggregation. This ultimately prevented the nuclear transport of SRRM2 and led to reduced levels of PQBP1 protein, which has been linked with the neurodevelopmental and intellectual disorders. The study results were reported in Molecular Psychiatry.
“We showed that the increased phosphorylation of SRRM2 prevented it from interacting with another protein which aids protein folding,” says first author Hikari Tanaka. “In the absence of this interaction, SRRM2 remained unfolded so was not transported to the nucleus and was degraded in the cytoplasm.”
The team next measured levels of SRRM2 and PQBP1 protein in the cerebral cortex of early-phase AD mice and human end-stage AD patients as well as in human AD iPS cells. Both proteins were greatly reduced compared with corresponding amounts in healthy controls.
“To find out what effect a reduction in PQBP1 would have in vivo, we generated knockout mice in which the PQBP1 gene was disrupted,” explains corresponding author Hitoshi Okazawa. “We observed cognitive decline and changes in the morphology of their synapses, which are junctions between neurons that allow electrical and chemical communication. These changes were caused by disrupted patterns of synapse gene splicing.”
A viral vector containing PQBP1 was used to recover the synapse protein expression in these mice. Not only did this restore PQBP1 expression, but it also recovered the abnormal phenotypes. These findings offer a new insight into early changes that occur during AD pathology involving splicing proteins, suggesting possibilities for gene therapies by virus vectors.
Tokyo, Japan – Alzheimer’s disease (AD) is the most common form of dementia, involving memory loss and a reduction in cognitive abilities. Patients with AD develop multiple abnormal protein structures in their brains that are thought to destroy or damage nerve cells (neurons). One of these structures, the senile plaque, is made up of clumps of beta-amyloid (Aβ) peptide which form in the spaces between neurons.
Many advanced clinical trials in patients with AD have attempted to slow down or reverse the disease by targeting these plaques for removal. However, despite the successful decrease in Aβ aggregation, these trials mostly have failed to improve memory or cognitive function in AD patients.
Before the formation of Aβ aggregates, studies revealed changes in the phosphorylation (a chemical modification) of certain proteins, including SRRM2. This protein was thought to be involved in a form of gene regulation known as splicing, but its exact function was unclear.
Now, a Japanese team of researchers led by Tokyo Medical and Dental University (TMDU) has examined levels of SRRM2 phosphorylation in a mouse model of AD, and found that they increased prior to Aβ aggregation. This ultimately prevented the nuclear transport of SRRM2 and led to reduced levels of PQBP1 protein, which has been linked with the neurodevelopmental and intellectual disorders. The study results were reported in Molecular Psychiatry.
“We showed that the increased phosphorylation of SRRM2 prevented it from interacting with another protein which aids protein folding,” says first author Hikari Tanaka. “In the absence of this interaction, SRRM2 remained unfolded so was not transported to the nucleus and was degraded in the cytoplasm.”
The team next measured levels of SRRM2 and PQBP1 protein in the cerebral cortex of early-phase AD mice and human end-stage AD patients as well as in human AD iPS cells. Both proteins were greatly reduced compared with corresponding amounts in healthy controls.
“To find out what effect a reduction in PQBP1 would have in vivo, we generated knockout mice in which the PQBP1 gene was disrupted,” explains corresponding author Hitoshi Okazawa. “We observed cognitive decline and changes in the morphology of their synapses, which are junctions between neurons that allow electrical and chemical communication. These changes were caused by disrupted patterns of synapse gene splicing.”
A viral vector containing PQBP1 was used to recover the synapse protein expression in these mice. Not only did this restore PQBP1 expression, but it also recovered the abnormal phenotypes. These findings offer a new insight into early changes that occur during AD pathology involving splicing proteins, suggesting possibilities for gene therapies by virus vectors.
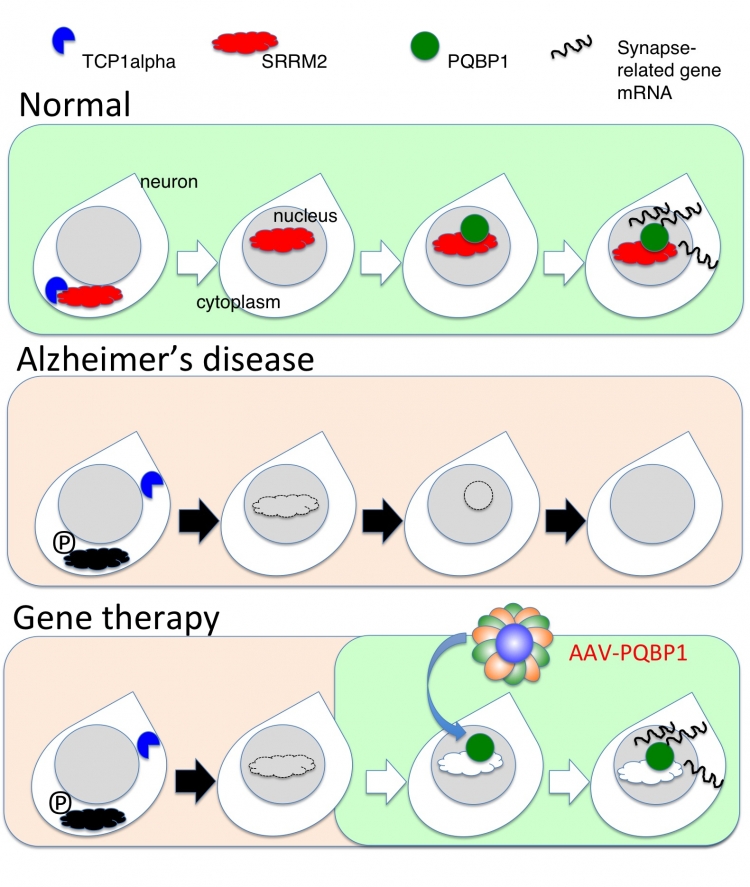
Theory of Gene Therapy by AAV-PQBP1: In Alzheimer’s disease state, the nuclear scaffold protein SRRM2 and the synapse-gene regulator PQBP1 are decreased. By increasing PQBP1 in neurons, expression of synapse genes are recovered and cognitive defects are rescued in Alzheimer’s patients.
The article, “The intellectual disability gene PQBP1 rescues Alzheimer’s disease pathology” was published in Molecular Psychiatry at DOI 10.1038/s41380-018-0253-8.
Summary:
TMDU-led Japanese research revealed a role for splicing proteins in the pathology of Alzheimer’s disease. Increased phosphorylation of the SRRM2 protein, seen in AD mouse models and human patients, was found to block its transport to the nucleus. This reduced levels of the PQBP1 protein, causing abnormal changes to the splicing of synapse genes and cognitive decline. These phenotypes were reversed by restoring PQBP1 function, suggesting a possible future treatment for AD.
Correspondence to:

Hitoshi OKAZAWA, MD, PhD, Professor
Department of Neuropathology,
Medical Research Institute,
Tokyo Medical and Dental University (TMDU)
E-mail: okazawa.npat(at)mri.tmd.ac.jp
Department of Neuropathology,
Medical Research Institute,
Tokyo Medical and Dental University (TMDU)
E-mail: okazawa.npat(at)mri.tmd.ac.jp