
“Clarification of a molecular mechanism underlying cerebellar degeneration”
Department of Neuropathology,
Division of Pathophysiology,
Medical Research Institute
Although several reports have proved implication of Glia cells in neurodegeneration, underlying detailed molecular mechanisms remained to be solved.
The group has identified the new molecule, MAXER though a comprehensive screening for identifying genes whose expression levels are altered in cerebellar cells of spinocerebellar ataxia type I (SCA1). The results indicate that MAXER levels are reduced only in SCA1 but not in another neurodegenerative disease such as Huntington disease. Biochemical analysis has revealed that MAXER is an evolutionary conserved endoplasmic reticulum-anchored membrane protein. Reduction of MAXER triggers cell cycle arrest at the G1 phase and MAXER regulates cell cycle through controlling a Cyclin D1 inhibitor, CDKRAP3. They indicated that reduction of MAXER in SCA1 causes reduction of Bergmann glia leading to increased glutamate toxicity in neurons resulting in neurodegenerative pathology.
This discovery is expected to be a clue for therapeutic development of neurodegenerative diseases by utilizing regeneration of Bergmann glia.
The research has been performed with Dr. Hiroki Shiwaku (a graduate student of Tokyo Medical and Dental University) and supported by Dr. Zoghbi, Baylor College of Medicine. This work has been on-line in EMBO Journal since June 8th, 2010.
This work has been supported by a research grant listed below.
Core Research for Evolutional Science and Technology (CREST)
Research field: “Development of novel technology for therapy and diagnosis of neurological and psychological disorder based on understanding of molecular pathology.”
Committee: Dr. Teruhiko Higuchi, President, National Center for Neurology and Phsychiatry.
Project title: Development of comprehensive therapeutic approaches of polyglutamine diseases.
Principal investigator: Dr. Nobuyuki Nukina, Group leader, RIKEN Brain Research Institute.
Co-investigator: Dr. Hitoshi Okazawa, Professor, Tokyo Medical and Dental University.
Project period: October, 2009-March, 2015
JST (Japan Science technology corporation) aims development of novel technologies by utilizing knowledge of basic brain science for prevention, therapy, and diagnosis of psychiatric and neurological disorders such as dementia and cognition disease, which are serious problems in Japan’s society along with falling birthrate and increase of aging population.
In this project, they seek for development of therapeutic approaches through inhibition of aggregation of abnormal protein and regulation of the protein degradation and of abnormal transcription in the pathological process in hereditary neurodegenerative disease, polyglutamine disease whose treatment is not established at all to date.
Research background
Spinocerebellar ataxia (SCA) is a hereditary neurodegenerative disorder whose population is the 3rd among neurodegenerative diseases following Alzheimer’s disease and Parkinson’s disease. Since there is no effective treatment, elucidation of the molecular pathology and development of therapeutic medicines have much attention in the society. SCA I has been known to have unusual extension of CAG repeat in the causative gene, Ataxin-1. Because CAG (C=cytosine; A=adenine; G=guanine) that codes an amino acid, glutamine is extremely extended also in other SCA (SCA2, SCA3, SCA6, SCA7, SCA17), they are referred as polyglutamine diseases and believed to have a common pathology along with Huntington disease and spino-bulbar muscular atrophy (SBMA).
In contrast, pathology of another neurodegenerative disease group, amyotrophic lateral sclerosis (ALS) has been implicated in events in glial cells, in which it is believed that astroglial cells of ALS transmit some toxic mediator to neurons, resulting in neurodegeneration. In addition, a study has revealed that Bergmann glia contribute to the neurodegeneration of SCA7 pathology. However, it is challenging problem to prove that non-cell autonomous pathology, which is neurodegenerative pathology triggered via glial cells is a common mechanism for all the neurodegenerative disorders. Furthermore, precise molecular mechanisms underlying neuronal cell degeneration have not been explained clearly. In SCA7, no molecular mechanisms implicated in Bergmann glia-mediated neurodegenerative pathology have been uncovered, and molecules that mediate a link between astroglia and neuronal degeneration has not been identified in ALS. Therefore, identification of a molecule that directly participates in glia-mediated neurodegeneration is expected in the society.
Research summary
A team in Tokyo Medical and Dental University led by professor Hitoshi Okazawa has performed comprehensive gene analysis in 3 types of primary neurons (Cortical, striatum, and cerebellar neurons. In cerebellar neuron culture, Bergmann glia are contaminated) expressing normal and mutant Huntington disease causative gene, mutant Huntingtin and SCA 1 causative gene, Ataxin-1 (ATX-1). In the analysis, the researchers sought to identify the genes whose expression levels in mutant Huntingtin or ATX-1 expressing cells are altered compared to the normal protein expressing cells (Tagawa et al., The Journal of Neuroscience, 2007). Among the 6 types of transcriptome data (normal and mutant ATX-1 in 3 cell-type,), a novel gene whose expression is reduced only in mutant ATX-1 expressing cerebellar neurons but not affected in other types of combination was identified. In this research project, the team has found that the novel molecule named “MAXER” is abundantly expressed in Bergmann glia of cerebellum (Fig. 1). Identification of the gene has uncovered a molecular mechanism underlying SCA1 pathology in which MAXER regulates proliferation of Bergmann glia. The researchers expressed MAXER protein fused with green fluorescent protein (GFP) to keep track the localization inside a cell and found that the protein colocalized with an endplasmic reticulum (ER) marker protein DsRed-ER. A biochemical analysis with ultra centrifugal separation of organelles also revealed that MAXER presents in the same fraction as an ER marker, indicating that MAXER is an ER protein. They also confirmed that the number of Bergmann glia was reduced in SCA1 pathology (Fig.2). Furthermore, depletion of MAXER molecule by RNAi method inhibited cell growth and inhibition of CDK5RAP3 that associates with MAXER accelerated cell growth.
The research has revealed that MAXER, a cell-proliferation regulating protein contributes to growth of Bergmann glia through an inhibition of CDK5RAP3 and that reduction of MAXER in SCA1 pathology creates an environment where Purkinje cell-protective Bergmann glia is significantly reduced causes neurodegeneration.
MAXER is only molecule to be shown that specifically regulates growth of Bergmann glia and has a role in the process of neurodegeneration mediated through glia to neurons (non-cell autonomous pathology) to date.
Implication of non-cell autonomous pathology in SCA1 is also a first example.
The study has been performed with a graduate student of Tokyo Medical and Dental University, Hiroki Shiwaku and supported by Dr. Zoghbi, Baylor College of Medicine for providing genetically engineered mice.
Future Prospective
The study has indicated importance of Bergmann glia in cerebellar degeneration. Several reports have proposed that Bergmann glia belong to stem cells since they maintain cell proliferation ability, which is different from neurons.
This discovery has provided a clue to be able to control proliferation of Bergnamm glia and increasing the number of Bergmann glia cells or improvement of function of Bergmann glia could contributes to novel therapeutic strategy for SCA.

Figure1. Maxer is expressed at Purkinje cell layer (purple) co-staining with anti-calbindin antibody (green, arrowheads). Maxer is expressed in calbindin-negative cells (arrows), but not in calbindin-positive Purkinje cells.
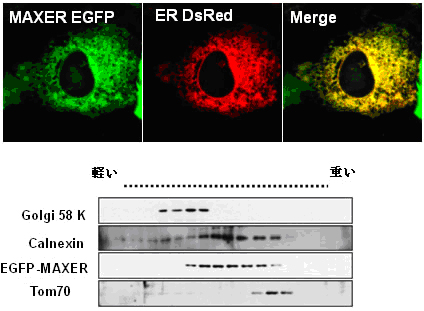
Figure 2. MAXER is an ER protein.
MAXER-EGFP colocalizes with ER-DsRED(Upper). Density gradient fractionation showed Maxer enrichment in ER, but not golgi or mitochondria. Calnexin (ER), golgi 58 K (golgi) and Tom70 (mitochondria) were used as organelle markers (lower).
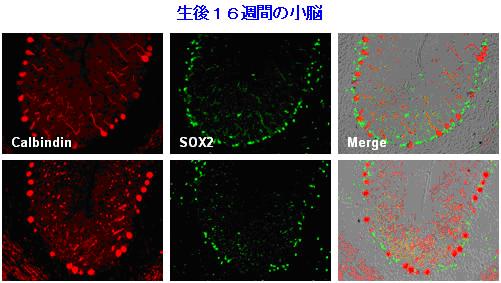
Figure 3. Bergmann glia were reduced in the cerebellum of mutant Atx1-KI mice. Bergmann glia and Purkinje cells were immunostained with anti-Sox2 antibody and anti-calbindin antibody, respectively. Corresponding cerebellar folia from 16-week old mice were analyzed.
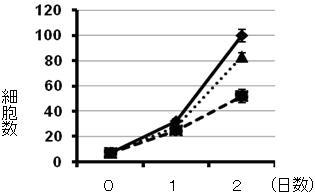
Figure 4. Effect of cell growth by MAXER. RNAi knock-down of MAXER inhibits cells growth (thicker dotted line) and additional RNAi knock-down of CDKRAP3 de-represses inhibition(thinner dotted line).
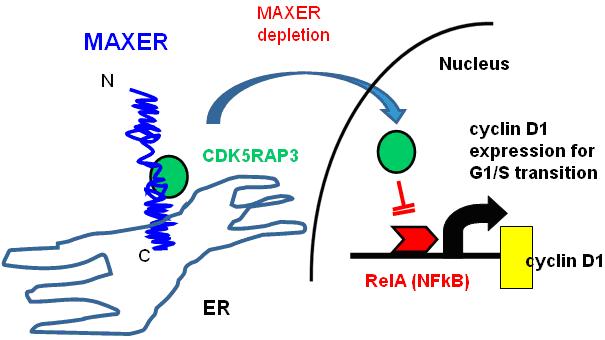
Figure 5. A hypothetical model of molecular function of Maxer to regulate cell cycle. MAXER (indicated in blue) tethers transcription regulatory molecule, CDK5RAP3 (indicated in green) that inhibits cell-cycle regulator, cyclin D1 (indicated in yellow) to the cytoplasm. Reduction of MAXER triggers translocation of CDK5RAP3 to the nucleus to inhibit RelA (NF-kB, indicated in red) that drives cyclin D1 transcription, resulting in cell cycle arrest in G1/S phase transition.
Key words
Bergmann glia
Bergmann glia are cerebellar astroglial cells that have their soma in the Purkinje cell layer and uptake glutamate as a neurotransmitter in the synapse cleft through AMPA receptor. Impairment of function of Bergmann gila leads to increase of local glutamate concentration, resulting in neurotoxicity.
Bergmann glia are suspected as stem cells because of its expression of neural stem cell specific protein, SOX2. However, the fact remains to be proven in terms of regeneration and development.
Spinocerebellar ataxia (SCA)
SCA is one of a group of brain diseases characterized by the degeneration of cerebellar neuron (Purkinje cells, the neuron of dentate nuclei etc). The degeneration of basal ganglia, retinal neuron or peripheral nerves is also accompanied in some cases.
There are hereditary SCAs and sporadic SCAs without clear family histories. The hereditary SCAs can be inherited in an autosomal dominant or autosomal recessive manner.
Endoplasmic reticulum (ER)
One of eukaryotic organelles: classified into rough ER that possesses ribosome and smooth ER that does not. In rough ER, translated proteins from mRNA on the ribosome are incorporated into the lumen of ER and managed for proper protein conformation, disulfide bond, and addition of sugar chains. When nascent proteins have unusual conformation, reconformation of the protein is executed. If the reconformation of protein fails, proteins are subjected for the degradation system called, ubiquitin-proteasome pathway.
Abnormal expansion of tri-nucleotide repeat
Polyglutamine disease including SCA1 is caused by increase of CAG repeat in the exon of the responsible gene from 20-30 to the pathological number more than 40 repeats. The repeats are believed to creates unusual expansion due to unstable in the process of DNA recombination and replication at the stage of gamete especially in spermatogenesis.
Polyglutamine diseases
Among the SCA, the group that shows symptoms by expansion of CAG repeats in the exon of responsible gene. Since CAG repeats in the exon code amino acid, glutamine, it leads to expansion of polyglutamine chain in the protein. SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, and DRPLA belong to polyglutamine disease. The abnormal proteins with expanded polyglutamine tract tend to form b-sheet structure that is suspected to contribute to protein aggregation.
Non-cell autonomous pathology
While cell autonomous pathology is cell death and functional impairment of neurons created by neuronal cells themselves, non-cell autonomous pathology is that the processes are mediated by abnormalities in other type cells such as glia cells.
Due to the secretion of toxic factor mediated by expression of degeneration causative gene product in astroglia cells, the model for non-cell autonomous pathology in which defect in neuroprotective glia, Bergmann glia cells contributes to neurotoxicity has been proposed.
Primary neuron culture
From brain of animals such as rat, cell culture is possible by segregation of cells with trypsin digestion followed by maintenance in the media containing serum and special ingredients. Primary neurons enable us to perform many kinds of experiments in neuroscience field.
Transcriptome data
Using small gene chips that cover all the genes from human, rat, and mouse, a comprehensive analysis of gene expression is possible. The data obtained in this gene analysis is called transcriptome data.
Published Paper
“Suppression of the novel ER protein MAXER by mutant ataxin-1 in Bergman glia contributes to non-cell autonomous toxicity”
Contact
| Hitoshi Okazawa, M.D., Ph.D. Department of Neuropathology, Division of Pathophysiology, Medical Research Institute, Tokyo Medical and Dental University Phone: +81-3-5803-5847 Fax: +81-3-5803-5847 E-mail: okazawa.npat@mri.tmd.ac.jp |