
老化と神経変性疾患の関係性を、核小体分子PQBP3が説明する
アルツハイマー病、パーキンソン病、脊髄小脳失調症、筋萎縮性硬化症などの神経変性疾患の多くは、中年期から老年期になって発症することから、老化が神経変性疾患の最大のリスク因子と考えられている。神経変性疾患は細胞内外の異常構造タンパク質蓄積と凝集を病理学的な特徴とするのに対して、老化はDNA損傷集積、テロメア短縮、酸化ストレスや終末糖化産物の集積、あるいは様々な酵素活性低下など複雑な側面を持っており、神経変性病態と老化病態の関係性は未だ十分に解明されているとはいえない。さらに、神経変性病態と老化病態のそれぞれの理解も完全ではないことから、両者の関係性の理解は生物学上そして医学上の重要課題として残っている。
また、老化のモデルとして「細胞老化(senescence)」が知られている。これは、培養細胞が分裂できる回数には上限があり(50回程度)、その結果として分裂を止めた細胞の状態を意味している。このような細胞老化では、細胞の核膜が不安定化していること、Lamin B1などの核膜の裏打ちタンパク質が減少していることが知られている。このような細胞老化は高齢者の皮膚や脳グリア細胞にも見られるため、senolysisと呼ばれる老化細胞除去による治療法の開発が進んでいる。しかし、もともと細胞分裂を停止している神経細胞であるニューロンに同様の治療法を用いることは、ニューロンを殺してしまうことになり、ニューロンにおける細胞老化と神経変性の関係性は、この点でも重要な研究課題である。
一方、私達の研究グループでは20年以上前に、神経変性疾患の主要カテゴリーのひとつであるポリグルタミン病(脊髄小脳失調症1型やハンチントン病を含む10種類以上の神経変性疾患が含まれる神経変性疾患グループ)の原因タンパク質と結合して細胞毒性の起点となる正常タンパク質を網羅的にスクリーニングを行い、この結果、PQBP1, PQBP3, PQBP5, VCPなどのタンパク質を発見した(Imafuku et al, BBRC 1998; Waragai et al, Hum Mol Genet 1999)。PQBP1はRNAスプライシングの調節因子であり、神経細胞ではシナプス関連遺伝子の発現調節に関わること(Okazawa et al, Neuron 2002; Ito et al, Mol Psychiatry 2015; Tanaka et al, Mol Psychiatry 2018)、遺伝性知的障害の原因遺伝子であること(Kalscheuer et al, Nature Genet 2003)、アルツハイマー病の神経細胞では減少していること(Tanaka et al, Mol Psychiatry 2018)、自然免疫細胞ではアルツハイマー病のタウ蛋白やエイズウィルス蛋白・DNAと結合して炎症反応を惹起すること(Yoh et al, Cell 2015; Yoh et al, Mol Cell 2022; Jin et al, Nature Comun 2021)、がわかっている。また、PQBP5は細胞の正常状態およびストレス状態において核小体構造を形成・維持するための必須分子であることがわかっている(Jin et al, Nature Commun 2023)。
本研究では、残されたPQBP3の機能解明を目指して、私達のグループが20年にわたって研究を続けた結果、老化と神経変性疾患の関係性における核小体分子PQBP3の役割が明らかとなった。
なお、PQBP3とPQBP5はこの20年の間に、他の研究グループが核小体構成成分の質量分析研究を行った際にNOL7, NOL10という名称をつけたため、PQBP3/NOL7あるいはPQBP5/NOL10とも呼ばれている。
また、老化のモデルとして「細胞老化(senescence)」が知られている。これは、培養細胞が分裂できる回数には上限があり(50回程度)、その結果として分裂を止めた細胞の状態を意味している。このような細胞老化では、細胞の核膜が不安定化していること、Lamin B1などの核膜の裏打ちタンパク質が減少していることが知られている。このような細胞老化は高齢者の皮膚や脳グリア細胞にも見られるため、senolysisと呼ばれる老化細胞除去による治療法の開発が進んでいる。しかし、もともと細胞分裂を停止している神経細胞であるニューロンに同様の治療法を用いることは、ニューロンを殺してしまうことになり、ニューロンにおける細胞老化と神経変性の関係性は、この点でも重要な研究課題である。
一方、私達の研究グループでは20年以上前に、神経変性疾患の主要カテゴリーのひとつであるポリグルタミン病(脊髄小脳失調症1型やハンチントン病を含む10種類以上の神経変性疾患が含まれる神経変性疾患グループ)の原因タンパク質と結合して細胞毒性の起点となる正常タンパク質を網羅的にスクリーニングを行い、この結果、PQBP1, PQBP3, PQBP5, VCPなどのタンパク質を発見した(Imafuku et al, BBRC 1998; Waragai et al, Hum Mol Genet 1999)。PQBP1はRNAスプライシングの調節因子であり、神経細胞ではシナプス関連遺伝子の発現調節に関わること(Okazawa et al, Neuron 2002; Ito et al, Mol Psychiatry 2015; Tanaka et al, Mol Psychiatry 2018)、遺伝性知的障害の原因遺伝子であること(Kalscheuer et al, Nature Genet 2003)、アルツハイマー病の神経細胞では減少していること(Tanaka et al, Mol Psychiatry 2018)、自然免疫細胞ではアルツハイマー病のタウ蛋白やエイズウィルス蛋白・DNAと結合して炎症反応を惹起すること(Yoh et al, Cell 2015; Yoh et al, Mol Cell 2022; Jin et al, Nature Comun 2021)、がわかっている。また、PQBP5は細胞の正常状態およびストレス状態において核小体構造を形成・維持するための必須分子であることがわかっている(Jin et al, Nature Commun 2023)。
本研究では、残されたPQBP3の機能解明を目指して、私達のグループが20年にわたって研究を続けた結果、老化と神経変性疾患の関係性における核小体分子PQBP3の役割が明らかとなった。
なお、PQBP3とPQBP5はこの20年の間に、他の研究グループが核小体構成成分の質量分析研究を行った際にNOL7, NOL10という名称をつけたため、PQBP3/NOL7あるいはPQBP5/NOL10とも呼ばれている。
本研究において、私達は、細胞老化においてはPQBP3が細胞核から減少すること、神経変性疾患病態においてはPQBP3が疾患タンパク質と結合して細胞核から減少すること、核膜の安定性を保つ機能を持つことを示し、ニューロンの細胞老化と変性病態がPQBP3機能低下という点で共通していることを最終的に示した。
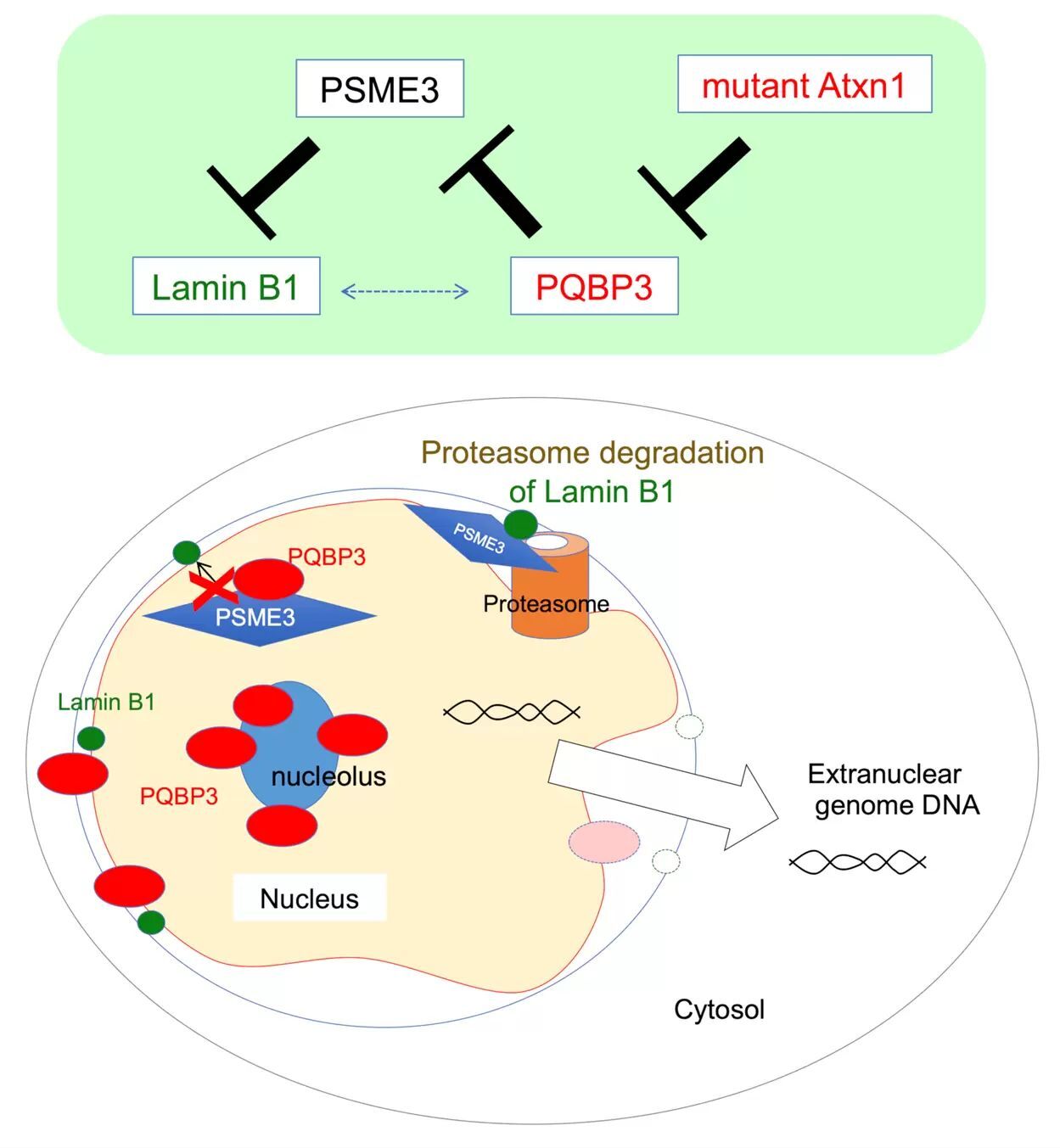
図1: PQBP3は核小体に貯蔵されているが、その一部が正常状態において核膜に移動し、PSME3による核膜裏打ちタンパク質Lamin B1の分解を抑制している。
細胞老化では、mTORシグナル活性化を通じてPQBP3が細胞質に移動してしまうために、核内および核膜裏打ち部から不足する。また、SCA1のようなポリグルタミン病では、疾患タンパク質が蓄積・凝集した封入体にPQBP3が引き寄せられるため、やはり、PQBP3が細胞質に移動してしまうために、核内および核膜裏打ち部から不足する。
PQBP3は通常は核内の核小体の周辺部に局在しているが、細胞が培養皿中で満杯状態になって細胞分裂を休止した時(quiescence)、あるいは、細胞培養で継代を繰り返したために細胞分裂が停止した時(senescence)では、核小体から核質(nucleoplasm)あるいは細胞質(cytoplasm)に移動していた。このPQBP3細胞内局在のシフトは、オートファジーシグナルにも関係するmTORによって影響を受けていた。さらに、過酸化水素によって細胞老化(senescence)の状態を誘導した際にも、PQBP3の細胞質シフトが起こった。また、この局在シフトには核DNAの細胞質への漏出を伴っていましたので、このようなPQBP3が細胞質に移動した細胞を電子顕微鏡で観察すると、核膜形態が不安定化していることがわかった。PQBP3の量をsiRNAによって減少させた場合にも、核DNAの細胞質漏出と核膜形態の不安定化が再現された。タンパク質間相互作用のデータベースを用いたバイオインフォマティクス解析から、PQBP3がタンパク質のプロテアソーム分解に関わるPSME3と結合することが示唆されたので、PQBP3・PSME3・核膜の3者の関係を詳細に検討したところ、正常状態において、一部のPQBP3は貯蔵基地としての核小体から離れて核膜に移動してPSME3に結合し、核膜裏打ちタンパク質であるLamin B1のSUMO化を介したタンパク質分解を抑えていること、細胞老化の状態では、これら一連の機能が低下すること、が明らかとなった(図1)。一方、ポリグルタミン病である脊髄小脳失調症1型(SCA1)の病態では、疾患タンパク質Ataxin-1の封入体にPQBP3が引き寄せられて核小体から減少し、核膜の不安定化が起きていることが示された。
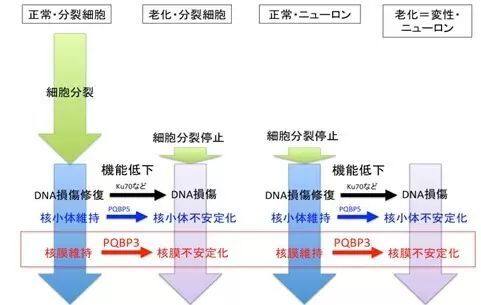
図2: 分裂細胞と非分裂細胞(ニューロン)のそれぞれにおける細胞老化の分子基盤を図示する。
本研究成果の重要性は、老化と神経変性の両病態に共通する新しい分子基盤を解明したことにある(図2)。PQBP3の機能低下は脳ニューロンにおいて細胞老化と神経変性の両病態で起きうるため、PQBP3を標的にすることで、脳老化と脳神経変性の両者を改善することが理論的には可能である。一方、ニューロン以外の分裂細胞では、PQBP3はガンのリスク因子として報告されており、これは細胞老化がガンを抑制するというガン研究領域の常識と一致する。したがって、PQBP3はいわば”諸刃の劍”であり、ガンと神経変性という対照的な細胞病態という生物学の根本的問題とも関わるため、ニューロン選択性の高い発現調節機構を利用するなどの技術的な工夫が必要と考えられる。
発表論文
Yoshioka, Y., Huang, Y., Jin, X., Ngo, K. X., Kumaki, T., Jin, M., Toyoda, S., Takayama, S., Inotsume, M., Fujita, K., Homma, H., Ando, T., Tanaka, H. & Okazawa, H. (2024)
PQBP3 prevents senescence by suppressing PSME3-mediated proteasomal Lamin B1 degradation.
The EMBO Journal 5 August 2024, 1–32. doi: 10.1038/s44318-024-00192-4
PQBP3 prevents senescence by suppressing PSME3-mediated proteasomal Lamin B1 degradation.
The EMBO Journal 5 August 2024, 1–32. doi: 10.1038/s44318-024-00192-4