
アルツハイマー病の発症前・超早期病態を部分的に解明 (2014)
アルツハイマー病の治療開発においては、発症前の早期病態を解明することが現在の最重要課題とされている。我々の研究グループは、最新の質量分析技術とスーパーコンピュータを用いたシステムズバイオロジーを駆使して、アルツハイマー病モデルマウスおよびアルツハイマー病患者脳のタンパク質を網羅的に解析し、発症前さらには老人班と呼ばれる異常タンパク質凝集が開始する前に、タンパク質リン酸化シグナルの異常が超早期病態として存在することを発見した。
アルツハイマー病は1902 年にドイツの病理学者アルツハイマーが初めて報告した進行性脳疾患であり、病理学的には脳組織の細胞外に老人班(Senile plaque)、細胞内に神経原線維変化(neurofibrillary tangle あるいは電子顕微鏡的にpaired helical filament) という2 つの特徴的な異常構造物が存在することが特徴である。前者はアミロイドベータ、後者はリン酸化タウというタンパク質の異常沈着であることが1980 年代の研究から明らかにされている。2000 年代以後、アミロイドベータの脳内凝集メカニズムをターゲットとする主として2 つの治療法が考案され、アルツハイマー病患者への臨床試験に至っている。その一つは、アミロイドベータに対する抗体を体内に投与して、アミロイドベータが脳内に凝集して老人班を形成することを抑制するものである。しかし、欧米の複数の巨大製薬会社がヒト臨床試験を行ったが、いずれも有効性を立証できなかった。特に重要なことは、死亡時まで追跡した患者の剖検脳において、抗体により老人班は消失していたにも関わらず、これらの患者の症状は改善していなかったという点である(Holmes et al., Lancet 2008)。また第2 の方法として、アミロイドベータを産生する酵素であるガンマセクレターゼの阻害剤のヒト臨床試験も巨大製薬会社によって行われたが、副作用によって開発中止に至っている(Doody et al., N Engl J Med. 2013)。これら既存のターゲットでは特効薬に至らなかった経緯を踏まえ、2014 年時点でのアルツハイマー病研究の最重要課題は、発症前あるいはアミロイドベータの凝集前の超早期病態を明らかにすること、さらには、そのような超早期病態に介入することで治療が可能であることを示すことにある。我々は14 年前にアミロイドベータが細胞外のみならず細胞内にも沈着すること、そのような沈着細胞ではc-Jun N-terminal kinase(JNK)というリン酸化酵素が活性化していることを報告している(Shoji et al., Molecular Brain Research 2000)。その後、アルツハイマー病以外の神経変性疾患を中心に網羅的質量分析(プロテオーム解析)を用いて、主要な病態分子を探索してきた(Qi et al., Nature Cell Biology 2007)。今回は最新の質量分析技術とスーパーコンピュータを用いたシステムズバイオロジーを駆使して、このようなアルツハイマー病における最重要課題の解決に取り組んだ。
今回の研究では、アルツハイマー病モデルマウス4 種類とアルツハイマー病患者の死後脳を対象に最新型の質量分析機を用いてプロテオーム解析を行った。さらに、東京大学ゲノム解析センターの宮野悟教授と共同研究を行い、プロテオーム解析で得られた膨大なデータをスーパーコンピュータで解析した。さらに、これらのビッグデータ解析から最終的に得られたコア分子を標的として、モデルマウスを用いた治療実験を行い、シナプスレベルの改善を認めた。ヒト死後脳では死後の保存条件によりタンパク質リン酸化が様々に変化することが知られている(Oka et al., PLoS ONE 2011)。そこで、今回の研究では、まずモデルマウス脳の質量分析とシステムズバイオロジー解析を始めに行い、中心的なリン酸化シグナルネットワーク異常を明らかにした後に、ヒト死後脳を用いて、コア病態ネットワークを確認するというストラテジーを採用した。モデルマウスは1種類ではなく4 種類を用いることで、個々の病態モデル特異的なバイアスを減らすことにした。対象としたのは、以下に示す家族性アルツハイマー病原因遺伝子の各変異を持つ4 種類のトランスジェニックマウスである。1)ヒトプレセニリン(PS)1 のエクソン9 欠損:PS1 マウス、2)ヒトPS2 のN141I 変異:PS2 マウス、3)ヒトアミロイド前駆体タンパク質(APP)のKM670/671NL(スェーデン型点変異):APP マウス、4)PS1(M146L,L285V 変異)とAPP(KM670/671NLスェーデン型変異、I716V フロリダ型変異、V717I ロンドン型変異)の5種類の変異:5XFAD マウス。これらのマウスの1ヶ月齢、3 ヶ月齢、6 ヶ月齢の大脳サンプルから信頼度95%で同定したリン酸化タンパク質のうち、コントロールマウスの値から変化が確認されたリン酸化タンパク質をリスト化した後に、これらを国立遺伝学研究所で公開しているタンパク質間結合データベース(PPI データベース)に重ね合わせることにより、それぞれのモデルマウスで成長・加齢とともに変化する異常リン酸化シグナルネットワークを描出した。
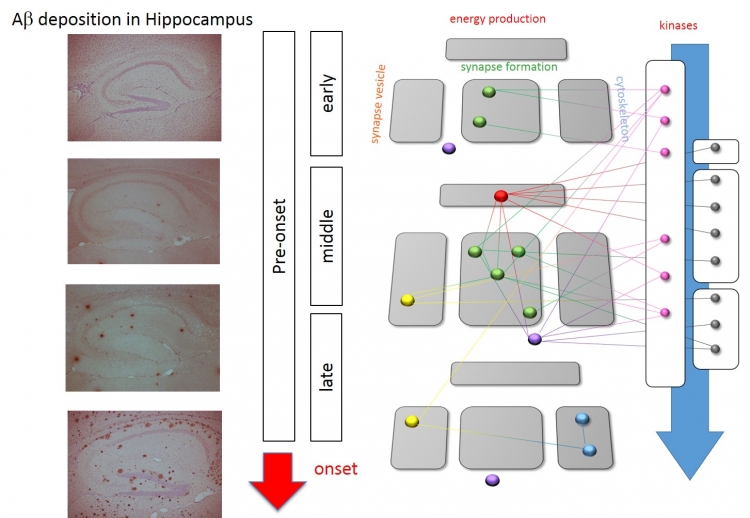
Phosphoproteomic changes before Amyloid deposition and Symptoms
さらに、アルツハイマー病のモデルマウス種を超えて共通する異常リン酸化シグナルネットワーク(コア病態ネットワーク)あるいはネットワーク構成タンパク質(コア病態タンパク質)は重要であるとの仮定のもとに、これらを抽出した結果、コア病態タンパク質はわずか17 個に集約しており、しかも、これらのタンパク質の大半が直接的に結合していることが明らかになった。さらに、コア病態タンパク質の大半はアルツハイマー病患者の死後脳でも変化が認められたこと、アミロイド病態の下流に位置づけられるタウ病態を反映すると考えられる変異型タウタンパク質を持つトランスジェニックマウスでの解析においても同様の変化が認められたことから、超早期から晩期に至るまでの持続的な異常シグナルであること、モデルマウスのみならずヒト病態にもトランスレータブルな変化であること、アミロイド病態とタウ病態をつなぐ変化であること、等の可能性が示唆された。分子機能面からコア病態ネットワークを眺めると、これらのタンパク質はシナプスに深く関わることが推測された。そこで最後に、4 種類のマウスのうち、最も症状の重篤な5XFAD マウスを用いて、治療実験を行った。近年、アルツハイマー病態においてシナプス異常が生じることが注目されている。我々の2 光子顕微鏡を用いた観察においても、5XFAD マウスの大脳皮質において興奮性シナプス後部の構造であるスパインが減少していることを確認した。
これまでに、同一研究室における4種類のモデルマウスに対しての同一手法による病理解析と行動解析の研究は行われていなかった。その実験を行った結果、もっとも重篤な5XFAD マウスでもアミロイド凝集が認められるのは3 ヶ月齢からであること、記憶力低下などの行動解析上の異常が検出されるのは6 ヶ月齢以後であることが分かった。したがって、コア病態ネットワークの変化は、発症前かつアミロイド凝集前であることが証明されたと考えられる。
コア病態ネットワークの中で最も早期からリン酸化が変化するコア病態分子は、MARCKS というリン酸化酵素protein kinase C (PKC) の基質であった。そこで、アデノ随伴ウイルス(AAV)を用いてスパインを蛍光標識し、PKC 阻害剤を5XFAD マウスに投与した後に2 光子顕微鏡で観察すると、スパイン減少が回復していることが明らかになった。またshRNA を用い、MARCKS 産生を減少させると、やはりスパイン減少が回復していることが示された。
以上の結果から、コア病態ネットワークの正しさが概ね証明されるとともに、コア病態タンパク質をターゲットとした治療開発が可能であることが示された。
コア病態ネットワークの中で最も早期からリン酸化が変化するコア病態分子は、MARCKS というリン酸化酵素protein kinase C (PKC) の基質であった。そこで、アデノ随伴ウイルス(AAV)を用いてスパインを蛍光標識し、PKC 阻害剤を5XFAD マウスに投与した後に2 光子顕微鏡で観察すると、スパイン減少が回復していることが明らかになった。またshRNA を用い、MARCKS 産生を減少させると、やはりスパイン減少が回復していることが示された。
以上の結果から、コア病態ネットワークの正しさが概ね証明されるとともに、コア病態タンパク質をターゲットとした治療開発が可能であることが示された。
本研究成果は、アルツハイマー病の発症前、凝集前の超早期病態の一端を捉えた成果と言える。明らかになった超早期のコア病態シグナルネットワークあるいはコア病態分子をターゲットとする治療法を本格的に開発することによってアルツハイマー病の進行を抑制し、治癒に導く治療法を開発できる可能性があると考えられる。
発表論文
Tagawa Kazuhiko, Homma Hidenori, Saito Ayumu, Fujita Kyota, Chen Xigui, Imoto Seiya, Oka Tsutomu, Ito Hikaru, Motoki Kazumi, Yoshida Chisato, Hatsuta Hiroyuki, Murayama Shigeo, Iwatsubo Takeshi, Miyano Satoru, Okazawa Hitoshi.
Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer’s disease brain.
Hum Mol Genet. 2015. 24 (2): 540-558. DOI: 10.1093/hmg/ddu475.
Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer’s disease brain.
Hum Mol Genet. 2015. 24 (2): 540-558. DOI: 10.1093/hmg/ddu475.