
「複数の神経変性疾患にまたがる共通病態(シグナル)を解明」【岡澤均 教授】
概要
アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症など神経細胞が死んでいく一連の疾患を神経変性疾患と呼びます。本研究は、これらの神経変性疾患は認知症、運動失調症、不随意運動など異なった症状を示すものの、TERA/VCP/p97という分子が共通して障害されていることを発見しました。TERA/VCP/p97分子を標的とすることで、幅広い適応疾患を持つ新しい治療法の開発につながることが期待されます。
背景
神経変性疾患には家族内発症者を持つ遺伝性のタイプと非遺伝性(孤発性)のタイプがあります。近年、遺伝性神経変性疾患の患者さんのサンプルを用いた分子遺伝学的解析から多くの原因遺伝子が見つかってきました。例えば、アルツハイマー病に次ぐ変性型認知症の原因疾患であり、前頭葉と側頭葉を強く障害する『前頭側頭葉変性症』(変性型初老期認知症の20%の原因と言われる)には、TDP43, PGN, TERA/VCP/p97, CHMP2B, C9orf72, FUSなどの原因遺伝子が過去5年ほどの間に発見されました。また、小脳を強く障害する神経変性疾患である脊髄小脳失調症の中には、さまざまな遺伝子において特定の塩基配列が異常に伸長しているケースが多いこと(グルタミンをコードしている塩基配列のために『ポリグルタミン病』※と総称されます)も明らかになってきました。これらのヒト原因遺伝子を組み込んだモデル動物(線虫、ショウジョウバエ、マウス、マーモセットなど)では、神経変性疾患類似の病態が再現されるために、ヒト神経変性疾患の分子病態解明と治療法開発に大きな貢献をしています。
一方、数多く存在する神経変性疾患が、お互いにどのような関係にあるのか、特に分子レベルの病態シグナル経路にどのような共通性があるのか、については良くわかっていません。また、それぞれの疾患の病態シグナル経路の特異性が、どのように疾患の症状の違いに結びつくのかについても十分に理解されていません。このような、疑問が解決すると、複数の神経変性疾患に同時に有効な治療薬・治療法、あるいは、特定の神経変性疾患に非常に有効な治療薬・治療法の開発に進むことが出来ると考えられています。
※ポリグルタミン病
DNA配列の中にグルタミンをコードする塩基のリピートが存在することがある。それによりグルタミン鎖(ポリグルタミン)が遺伝子産物に含まれることになるが、ポリグルタミン病は、このポリグルタミンの毒性によって発症する。現在、ハンチントン病、球脊髄性筋萎縮症、遺伝性の脊髄小脳失調症、DRPLA(歯状核赤核淡蒼球ルイ体萎縮症)などが、ポリグルタミン病として知られている。
研究内容
この研究は、岡澤教授の研究グループの15年前の発見に端を発しています。同グループはポリグルタミン病原因タンパク質に結合する正常タンパク質を探索していました。この結果、PQBP1(その後、発達障害原因遺伝子であることも判明)とともにTERA/VCP/p97 (以下、VCPと記述) がポリグルタミン配列に結合することを見つけました(Imafuku et al, Biochem Biophys Res Commun 1998)。その後、他の研究グループもAtaxin-3というポリグルタミン病原因タンパク質の一つがVCPに結合することを示しました(Hirabayashi et al, Cell Death Differ 2001)。この結果から、VCPこそがポリグルタミン病の病態に直結した分子ではないかという仮説を立てました。
そこで、この仮説をさらに検証し、さらに一般化するために、他のポリグルタミン病の原因遺伝子についてVCPとの結合を検討しました。Ataxin-1(脊髄小脳失調症1型の原因遺伝子)、Ataxin-7(脊髄小脳失調症7型の原因遺伝子)、アンドロジェン受容体(球脊髄性筋萎縮症の原因遺伝子)、ハンチンチン(ハンチントン病の原因遺伝子)という、4種類のポリグルタミン病の疾患遺伝子を用いて検討したところ、正常型、変異型ともにポリグルタミン病タンパク質はVCPに結合し、ポリグルタミン病タンパク質からポリグルタミン配列だけを除いた変異体タンパク質とは結合しませんでした。このことから、ポリグルタミン病原因タンパク質とVCPの結合は一般化できることが示されました。
次に研究グループは、VCPにポリグルタミン病原因タンパク質が結合することによる細胞内部の変化について解析しました。VCPは「膜輸送」「小胞体関連タンパク質分解」「DNA損傷修復」などの様々な細胞機能に必要なエネルギーを供給する役割があることが知られています。そこで、複数のポリグルタミン病モデル動物および培養細胞を用いて、この3つの細胞機能について検討しました。変異ポリグルタミン病タンパク質は、非神経細胞において「膜輸送」や「小胞体関連タンパク質分解」を阻害するものの、培養神経細胞ではこれらの機能阻害は軽度でした。一方、神経細胞でも非神経細胞でも、ともに変異ポリグルタミン病タンパク質は、VCPを凝集体に引き込み、VCPの「DNA損傷修復機能」を阻害しました。実際に、マウス脳組織の免疫染色では、VCPは神経細胞においては核に主に存在しており、DNA損傷修復機能に主に関与することが示唆されました。以上の実験結果から、ポリグルタミン病原因タンパク質は、VCPのDNA損傷修復機能を阻害して神経変性につながっていることが明らかになりました。さらに、正常VCPを補充することにより、疾患モデル動物(ショウジョウバエ)において神経細胞のDNA損傷が軽減し、寿命が延長することも明らかになりました。
前述したように、ヒトVCP遺伝子変異そのものによって前頭側頭葉変性症を発症することが知られています。この変異によってVCPの機能が亢進するのか低下するのかについてはよく分かっていませんでした。例えば大腸菌で作った変異VCPタンパク質ではATP分解酵素活性が上昇しているとの報告もありました(Halawani et al, Mol Cell Biol 2009)。しかし、その後、VCP遺伝子変異はミトコンドリアの機能障害を介してATP産生の減少につながること(Baltolome et al, Neuron 2013)など、遺伝子変異はVCPの機能低下につながるという報告が増えています。したがって、本研究で示した4種類のポリグルタミン病におけるVCPの機能低下という所見と合わせると、ポリグルタミン病と前頭側頭葉変性症は、VCP機能という点から病態を共有することを意味しています。
今後の展望
また、岡澤グループは、文部科学省脳科学研究戦略推進プログラム課題E「心身の健康を維持する脳の分子基盤と環境因子」において、網羅的な病態シグナル解析を行っています。本研究成果と網羅的シグナル解析結果とを参照することで、環境因子がどのように前頭側頭葉変性症やポリグルタミン病などの認知症に影響を与えるかについても、今後の成果が期待できます。
本成果はNature Communications誌オンライン版(米国東部時間2013 年5月7日発行)に掲載されました。本研究は、東京医科歯科大学と、カリフォルニア大学サンディエゴ校、セント・ジュード小児病院、名古屋大学、マックスデルブルック分子医学研究所などとの共同で行われました。また本研究は、文部科学省脳科学研究戦略推進プログラム課題E「心身の健康を維持する脳の分子基盤と環境因子」、文部科学省新学術領域研究「シナプス・ニューロサーキットパソロジーの創成」、JST戦略的創造研究推進事業CREST「精神・神経疾患の分子病態理解に基づく診断・治療へ向けた新技術の創出」の支援により実施されました。
論文タイトル
(「複数のポリグルタミン病におけるDNA損傷修復障害にTERA/VCP/p97が寄与する」)
お問い合わせ先
岡澤 均
Tel: 03-5803-5847, FAX: 03-5803-5847, Email: okazawa.npat(ここに@を入れてください)mri.tmd.ac.jp
東京医科歯科大学 広報部広報課広報掛
〒113-8510 東京都文京区湯島1-5-45
Tel: 03- 5803-5833, FAX: 03-5803-0272, Email: kouhou.adm(ここに@を入れてください)tmd.ac.jp
文部科学省 脳科学研究戦略推進プログラムについてのお問い合せ
脳科学研究戦略推進プログラム 事務局
担当:大塩立華
TEL: 03-5282-5145, FAX: 03-5282-5146, Email: srpbs(ここに@を入れてください)nips.ac.jp
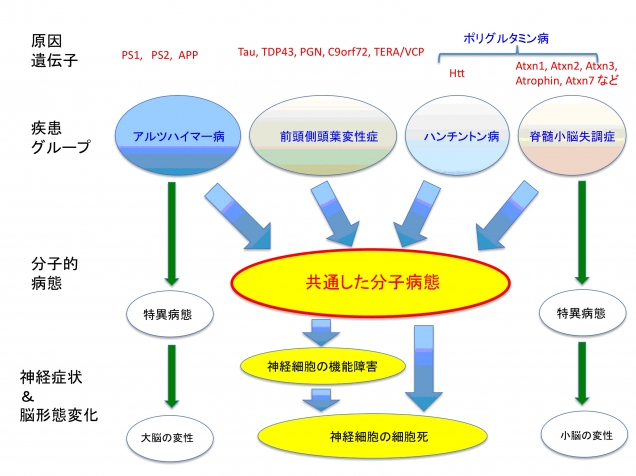
図1. 実験の概要。神経変性疾患には複数のグループがあり、異なる原因遺伝子を持っている。例えば、遺伝性アルツハイマー病の原因遺伝子はPS1, PS2, APPなどがあるが、前頭側頭葉変性症はTau, TDP43, TERA/VCPなどである。ハンチントン病と脊髄小脳変性症の5種類は原因遺伝子産物内のポリグルタミン配列が伸長して起きることからポリグルタミン病と総称される。これらの疾患には共通した分子病態と個別の分子病態があると仮定できる。今回の研究は、前頭側葉変性症、ハンチントン病、脊髄小脳失調症にまたがる共通病態としてTERA/VCP/p97機能低下があることを示した。
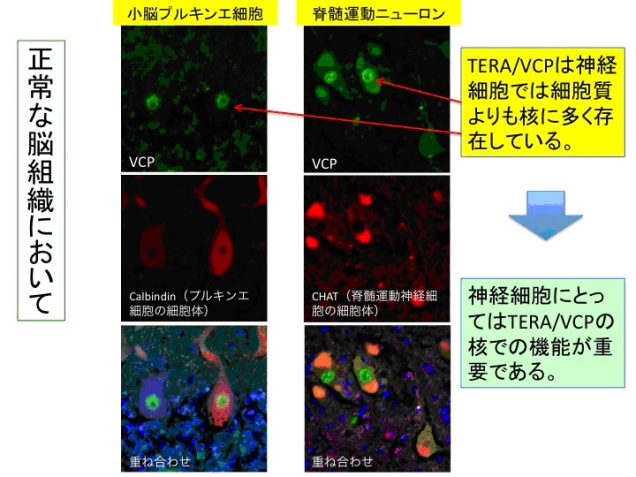
図2.TERA/VCP/p97タンパク質タンパク質は神経細胞の核に優位に存在している。
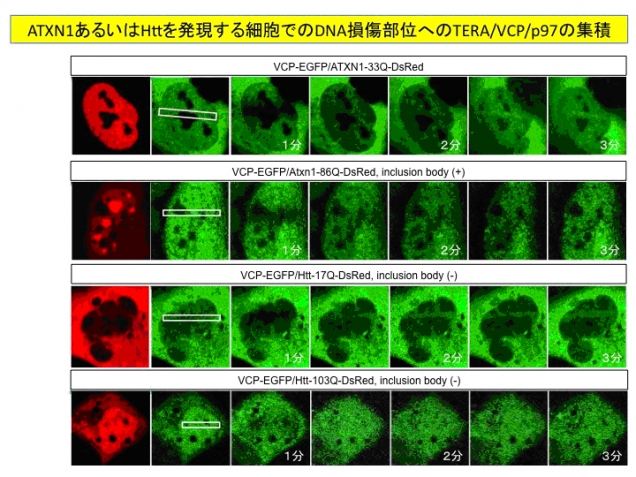
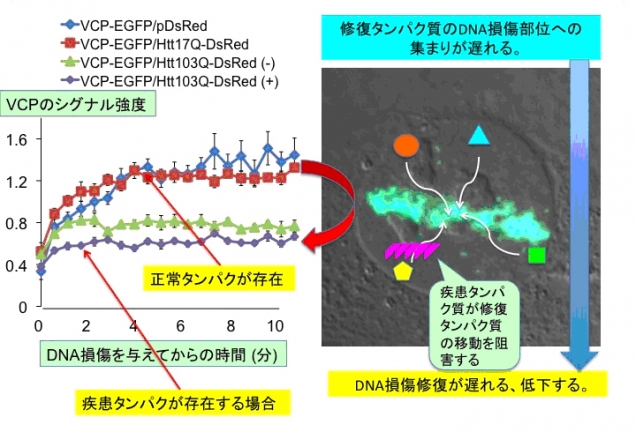
図3. ポリグルタミン病タンパク質はDNA損傷修復のための核内TERA/VCP/p97タンパク質のDNA損傷部への移動を抑制する。
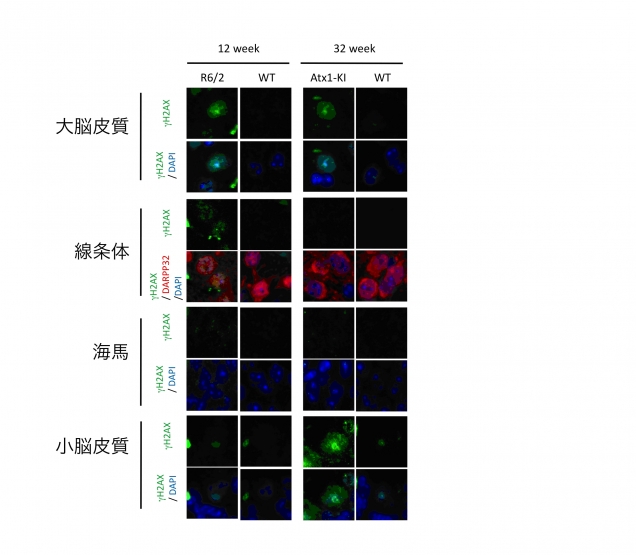
図4. ポリグルタミン病では一般的にDNA損傷が増加する。この図では、ハンチントン病モデルマウス(R6/2)と、脊髄小脳失調症モデルマウス(Atx1-KI)を示している。DNA損傷マーカー(γH2AX)は正常マウス(WT)に比べて、R6/2とAtx1-KIの神経細胞内で増加している。
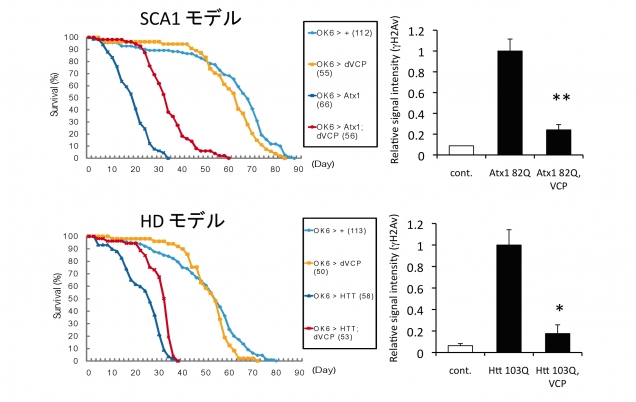
図5. TERA/VCP/p97を神経細胞に発現させるとDNA損傷修復を介してポリグルタミン病動物モデル(この場合はショウジョウバエ)の寿命を延長する。左図は、モデルショウジョウバエの神経細胞にTERA/VCP/p97を併せて過剰発現させると(赤線)、疾患モデルショウジョウバエ(SCA1:脊髄小脳失調症1型、HD:ハンチントン病)(濃青線)に比べて寿命が延長することを示す。右図は同じショウジョウバエでのDNA損傷の定量を示す。モデルショウジョウバエ(中間の棒)に比べて、TERA/VCP/p97過剰発現ショウジョウバエではDNA損傷が減少している。
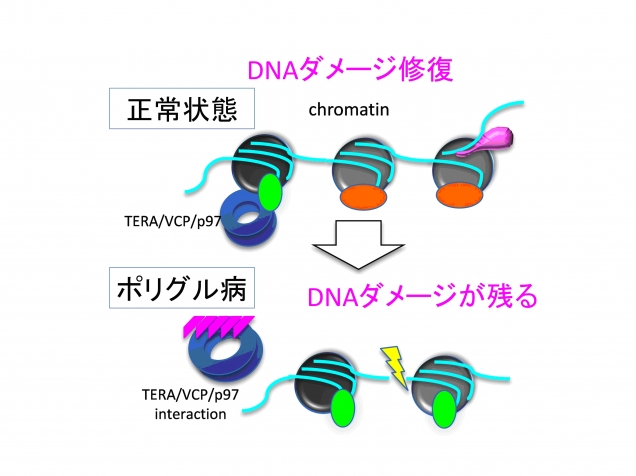
図6. ポリグルタミン病では一般にTERA/VCP/p97の核内動態が抑制されてDNA損傷修復が妨げられる。TERA/VCP/p97機能障害は前頭側頭葉変性症でも共通している。