
藤吉 好則:主な研究結果の解説
- 1.はじめに(分子構造の観察と電子線損傷)
- 2.クライオ電子顕微鏡の開発
- 3.電子線結晶学による膜タンパク質の構造解析
- 4.クライオ電子顕微鏡によるウイルス像の観察
- 5.バクテリオロドプシンの構造解析
- 6.水チャネル(電子線結晶学)
- 7.イオンチャネル
- 8.アセチルコリン受容体
- 9.ギャップジャンクションチャネル
- 10.タイトジャンクション
- 11.Gタンパク質共役型受容体(X線結晶学)
- 12.単粒子解析法とクライオ電子顕微鏡システム
- 13.水チャネル(単粒子解析法)
- 14.RNA標的創薬
- 15.Gタンパク質共役型受容体(単粒子解析)と構造創薬
- 16.構造創薬を活用した新型コロナウイルス治療薬候補の開発
→細胞構造生理学研究室ホームページ
→藤吉好則:主な研究結果の解説をPDFで見る
→藤吉好則:主要な研究業績をPDFで見る
1.はじめに(分子構造の観察と電子線損傷)

図1 加速電圧500kVの電子顕微鏡で撮影した塩化フタロシアニン銅の分子像。

図2 MDSで撮影したAg-TCNQの分子像。右下に構造を表示。
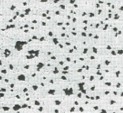
図3 気液界面膜の暗視野像。脂質分子が存在する部分が白く見え、黒い部分は穴である。
2.クライオ電子顕微鏡の開発
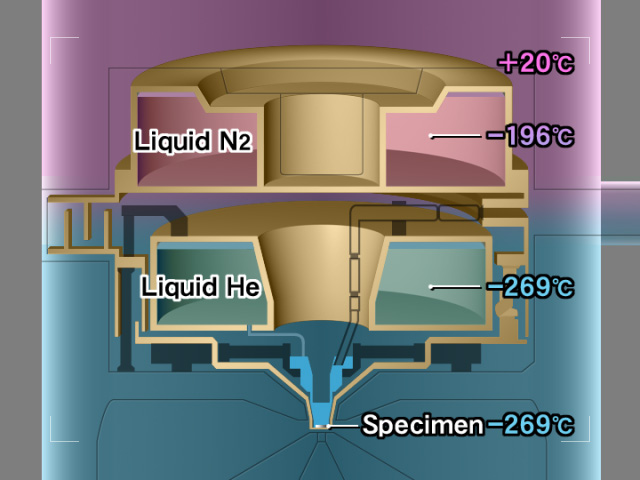
図4 低温ステージの模式図。液体ヘリウムタンクは完全に液体窒素で冷却された金メッキした銅板でシールされている。

図5 クライオ電子顕微鏡用ステージ。液体窒素タンクを外した状態で液体ヘリウムタンクとポットが見える。

図6 クライオ電子顕微鏡開発の推移。1983年から開発を開始して、1986年に最初のクライオ電子顕微鏡の開発に成功してから、改良を重ねてきた。第8世代のクライオ電子顕微鏡(JEM-Z320FHC)を2020年に開発して活用している。
3.電子線結晶学による膜タンパク質の構造解析
図7 電子線結晶学で構造解析された光合成アンテナタンパク質の構造。膜面に水平方向から見た構造。
4.クライオ電子顕微鏡によるウイルス像の観察

図8 クライオ電子顕微鏡で撮影したインフルエンザウイルス像。脂質2重膜構造ではない。
図9 インフルエンザウイルスの構造モデル。エンベロープは脂質2重膜ではなく、脂質1重膜に裏打ち構造からなる。
5.バクテリオロドプシンの構造解析
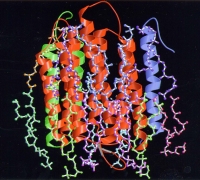
図10 クライオ電子顕微鏡と電子線結晶学を用いて解析したバクテリロドプシンと脂質分子の構造。
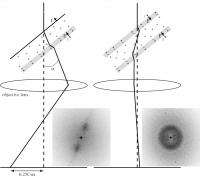
図11 チャージアップによる像のシフトとcarbon sandwich法による問題改善を示す模式図と実際の結果。
6.水チャネル(電子線結晶学)

図12 Engel等と共同で電子線結晶学により解析したAQP1の右巻ヘリカルバンドル構造。
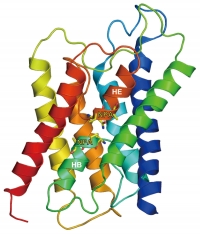
図13 電子線結晶学で解析したヒトAQP1の構造。
この構造解析結果を基に、水チャネルの速い水透過と高い水選択性の機能を理解するモデル、Hydrogen-Bond Isolation Mechanismを提案した(Nature, 407, 599-605, 2000)。その結果、K+イオンチャネルのイオン選択性の機構を解明したMacKinnonとともに、共同研究者Agreが2003年のノーベル化学賞を受賞した。しかし、このAQP1の構造は分解能が3.8Åと低く、チャネル内の水分子を観察できていなかったので、この時点ではHydrogen-Bond Isolation Mechanismはチャネルの構造から考案した単なる仮説に過ぎなかった。

図14 電子線結晶学で1.9 Å分解能で解析したAQP0の構造。脂質分子も観える。
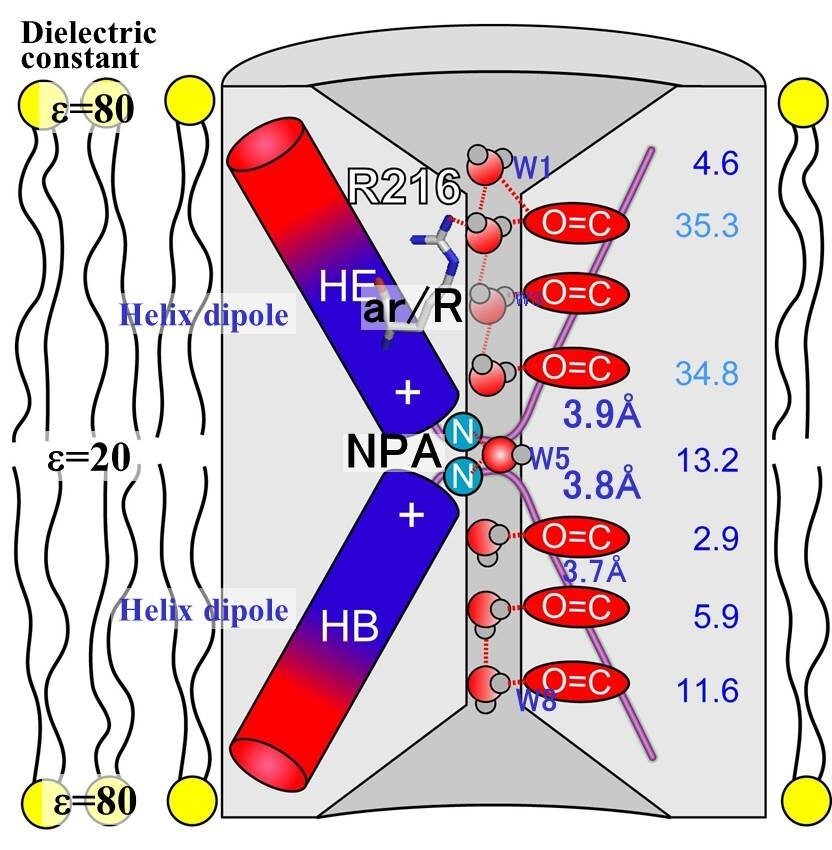
図15 水チャネルの2本の短いへリックスが形成するへリックス双極子によって、チャネル内の水分子が配向され、それと呼応する位置にカルボニル基が配置され、チャネル内の水分子はカルボニル基と水素結合を形成することによって、8か所にある水分子が観察された。
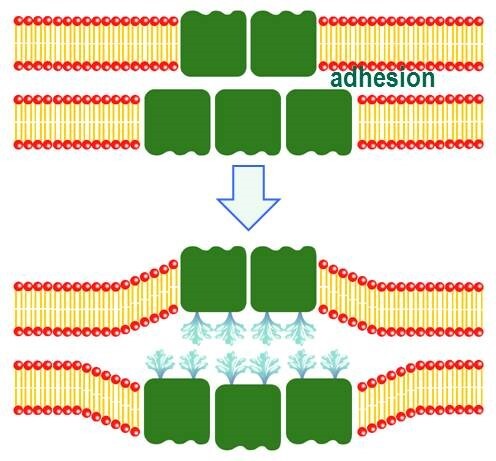
図16 浸透圧や水透過のセンシングをAQP4の弱い接着によって行っている可能性を示すモデル図。
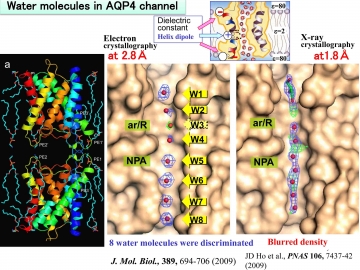
図17 電子線結晶学では、脂質膜の中で構造解析できているが、X線結晶学では脂質分子が除かれているために、短いヘリックスの静電場が弱くて、水分子が配向できていないために水分子が入りやすい位置が形成されていない。
脂質膜内で構造解析する電子線結晶学では、生理的条件に近い状態での解析ができているので水分子が配向し、その配向に呼応する位置にカルボニル基が水素結合を形成できるように配置されて、水分子が存在しやすい8個の位置がチャネル内に形成されている(図15、図17)。
一方、脂質膜が無いX線結晶学での解析では、この静電場が弱くて水分子を配向できないことによって水分子が存在しやすい位置がチャネル内に形成されないために、密度図がボケていると解釈される(図17参照)。
これまでに構造解析された全てのチャネルには短いヘリックス構造が観られている。それゆえ、脂質膜内においてヘリックス双極子が形成する静電場が、いろいろなチャネルにおいても重要な生理機能を担っていることが示唆される(Review: Proc. Jpn Acad., Ser B., 91, 447-468, 2015)(図18)。少なくとも、カチオンチャネルは短いヘリックスのC末端側がカチオンの入る位置に向いており、イオンがチャネル内に入りやすくしており、アニオンチャネルでは、N末端側がアニオンを入りやすくしていると考えられる。
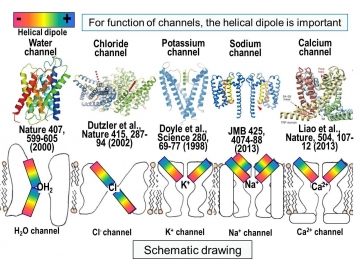
図18 水チャネルとイオンチャネルに見られる短いへリックスのヘリカル双極子の重要性を示唆する例。
7.イオンチャネル
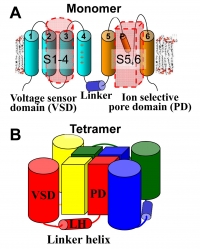
図19 バクテリア由来の電位感受性Na+チャネルは6回膜貫通ヘリックス構造を有しており(A)、それが4量体を形成している(B)。ただし、ポアドメインと電位感受性ドメインは1つ離れた隣に配置している。
電位感受性Na+チャネルのinactivationの機構を理解するために、このタイプのチャネルのC末端部分の構造解析と、変異体の構造情報と電気生理学的測定の結果から、C末端部分に形成されるヘリカルバンドルの安定性がinactivationの速さを制御していることを解明した(Nat. Commun., 3, 793, 2012)(図20)。
また、電位感受性Na+チャネルの2つの状態の構造を電子線結晶学で解析することによって、ゲーティング機構の一端が理解できるようになってきた(J. Mol. Biol., 425, 4074-4088, 2013)(図21)。
ゲーティング機構を理解するためにはresting stateの構造が必要であるが、膜電位が存在する状態での構造解析は困難であった。しかし、R. MacKinnon等はこのような解析をproteoliposomeの中にKV7.1 (KCNQ1)が存在する状態で解析して、見事に膜電子がかかった状態の構造を解析できるようにした(VS Mandala, R MacKInnon, PNAS, 120, 2301985120, 2023)。この様な方法により、電位感受性のゲーティング機構が詳細に理解できるようになる可能性があり興味深い。
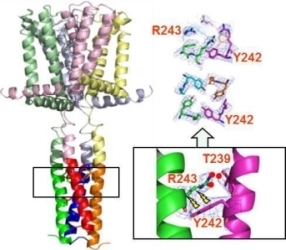
図20 C末端が形成する4本のヘリカルバンドルがinactivationに関わっており、それが安定なほどinactivationが速くなっている。
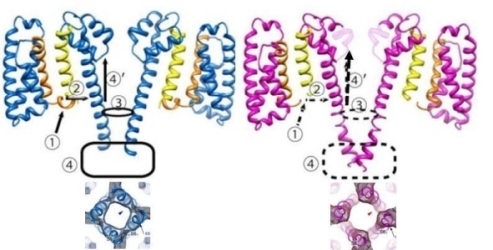
図21 Na+チャネルの2つのコンフォメーションの構造。
8.アセチルコリン受容体

図22 アセチルコリン受容体のチューブ状結晶を撮影したフィルム。G3 cryo-EM。
図23 アセチルコリン受容体のゲーティングモデル。
9.ギャップジャンクションチャネル
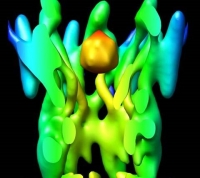
図24 電子線結晶学で解析したコネキシン-26のM34A変異体の構造。
さらに、X線結晶学を用いてワイルドタイプのコネキシン-26の構造が解析され、原子モデルが提案された(Nature, 458, 597-602, 2009)(図25)。
組織内でのギャップジャンクションチャネルの構造を解析するために、Lateral giant fiberの電気シナプスの構造を電子線トモグラフィー法で解析して、ギャップジャンクションが近傍に多く存在するベシクルと連結している構造を観察した(J. Struct. Biol., 175, 49-61, 2011)(図26)。
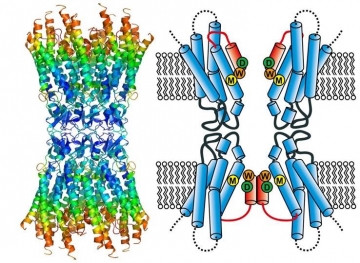
図25 コネキシン-26の構造とチャネル構造の模式図。
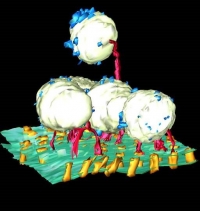
図26 電子線トモグラフィーで解析したベシクルを含むギャップジャンクションの立体的構造。
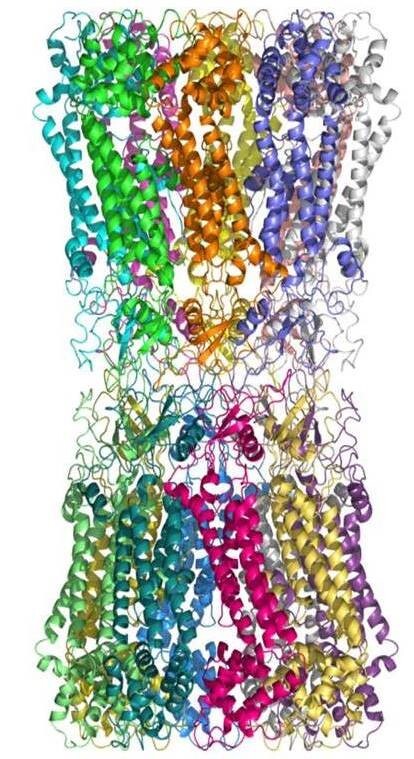
図27 クライオ電子顕微鏡と単粒子解析法を用いて解析したイネキシン-6が形成するギャップジャンクションの構造解析結果。結晶学では解析できなかった細胞質側の構造も構造モデルが作製出来る構造解析が可能であった。
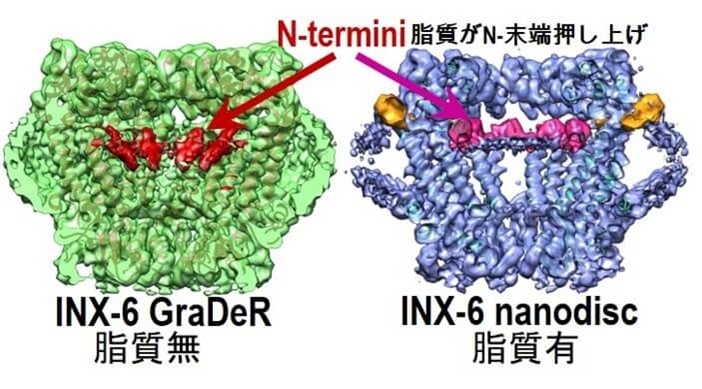
図28 Innexinの界面活性剤中での構造(左)とnanodiscでの構造解析(右)。脂質膜中ではチャネル内に脂質分子が入ってInnexinのN末端部分を押し上げている。
例えば、イネキシンの開閉構造を模式的に示す図30のように、lipid mediated gating mechanismを提案することができる。このギャップジャンクションチャネルファミリーの様に、大きなチャネル径を有する場合には、脂質分子が寄与する極めて興味深く重要なイオンチャネルのゲーティング機構が考えられる(Sci. Adv., 6, eaax3157, 2020, Sci. Signal., 15, eabg6941, 2022)。
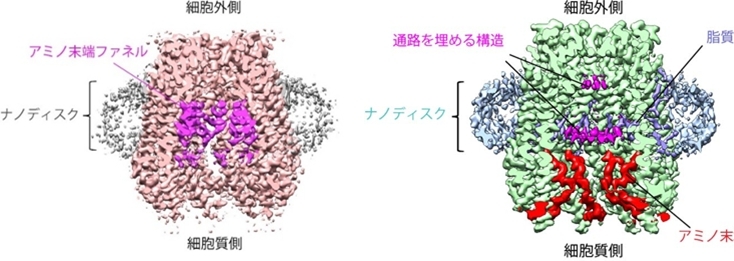
図29 Pannexinのnanodiscでの構造解析。Probenecid非結合(右)と結合(左)の構造。この記憶改善薬から痛風薬などの幅広い効果が知られているProbenecidによって、脂質が膜内に入ってチャネルを閉じている構造が解析された。
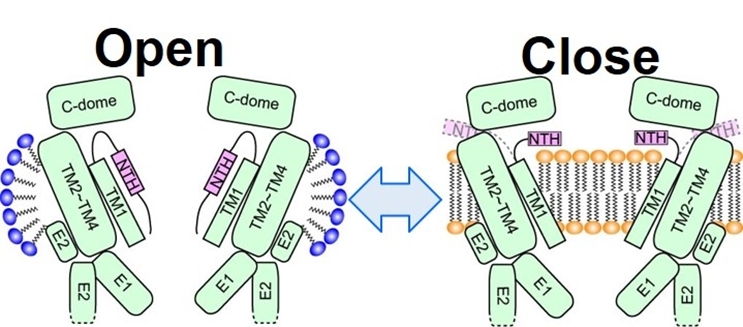
図30 Innexinについての脂質分子が関与するgating model: lipid mediated gating mechanism。
10.タイトジャンクション
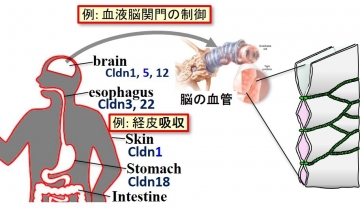
図31 タイトジャンクション(TJ)によるバリア形成を赤色の線で示す。血液脳関門とそれを形成するTJの拡大した模式図を右に示す。
10.1 タイトジャンクションとクローディン
我々の身体は、上皮細胞などによって外界と仕切られており、器官などのコンパートメントも仕切られている(図31)。それゆえ、上皮細胞間は、タイトジャンクション(TJ)と呼ばれるバリアによってシールされている。脳における血液脳関門(BBB: Blood Brain Barrier)では、このTJが形成され脳に異物が入るのを防ぐ重要なバリア機能を担っている。ただし、このTJは単にバリアとしてだけではなく、パラセルラーチャネルとして陽イオンや陰イオンなどの選択的な透過制御機構をも担っている。このTJを形成し、その性質の多様性を実現している中心的分子はクローディン(Cldn)である。ヒトのCldnsは27種類が発現していることが知られており、Cldnが存在する器官や組織は特徴的であり、TJのストランドはその数も多様である。すなわち、Cldnの種類などによって複数のストランドからなるTJが形成されている場合が多い(図31)。TJは27種類のCldnsや、それらと結合する足場タンパク質によってその性質が決定されている。この様な、Cldnsを中心とするTJに関する研究は、月田早智子博士らと一貫した共同研究として進めている。
10.2 クローディンの構造解析
X線結晶学を用いることで、Cldn15の構造を解析することに初めて成功した(Science, 344, 304-307, 2014)(図32)。その結果、クローディンは、4本の左巻きのヘリカルバンドル構造を形成し、細胞外のECS1とECS2が5本のβシート構造を形成していることを明らかにした。この構造は左手の手のひらに似ているので、手のひら構造モデルを提案した(図32)。
10.3 タイトジャンクションの構造モデル
結晶学的手法で構造解析した、このCldn15の結晶内相互作用構造も参考にして、TJの構造モデルとして、anti-parallel double row modelを提案した(J. Mol. Biol., 427, 291-297, 2015)(図33)。
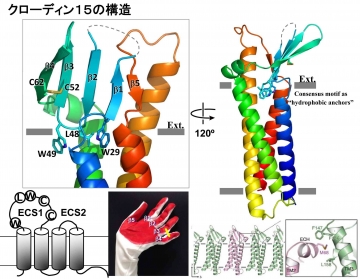
図32 クローディンの構造。4本の膜貫通ヘリックスが左巻きのヘリカルバンドルを形成し、細胞外で5本のβシート構造を形成している。それゆえ、手のひらモデルを提案した。またTJストランドを形成する相互作用様式を示唆される結果を得た。

図33 クローディン‐15の構造を基に作製したTJのダブルロウモデル。パラセルラーチャネル機能も理解できる。
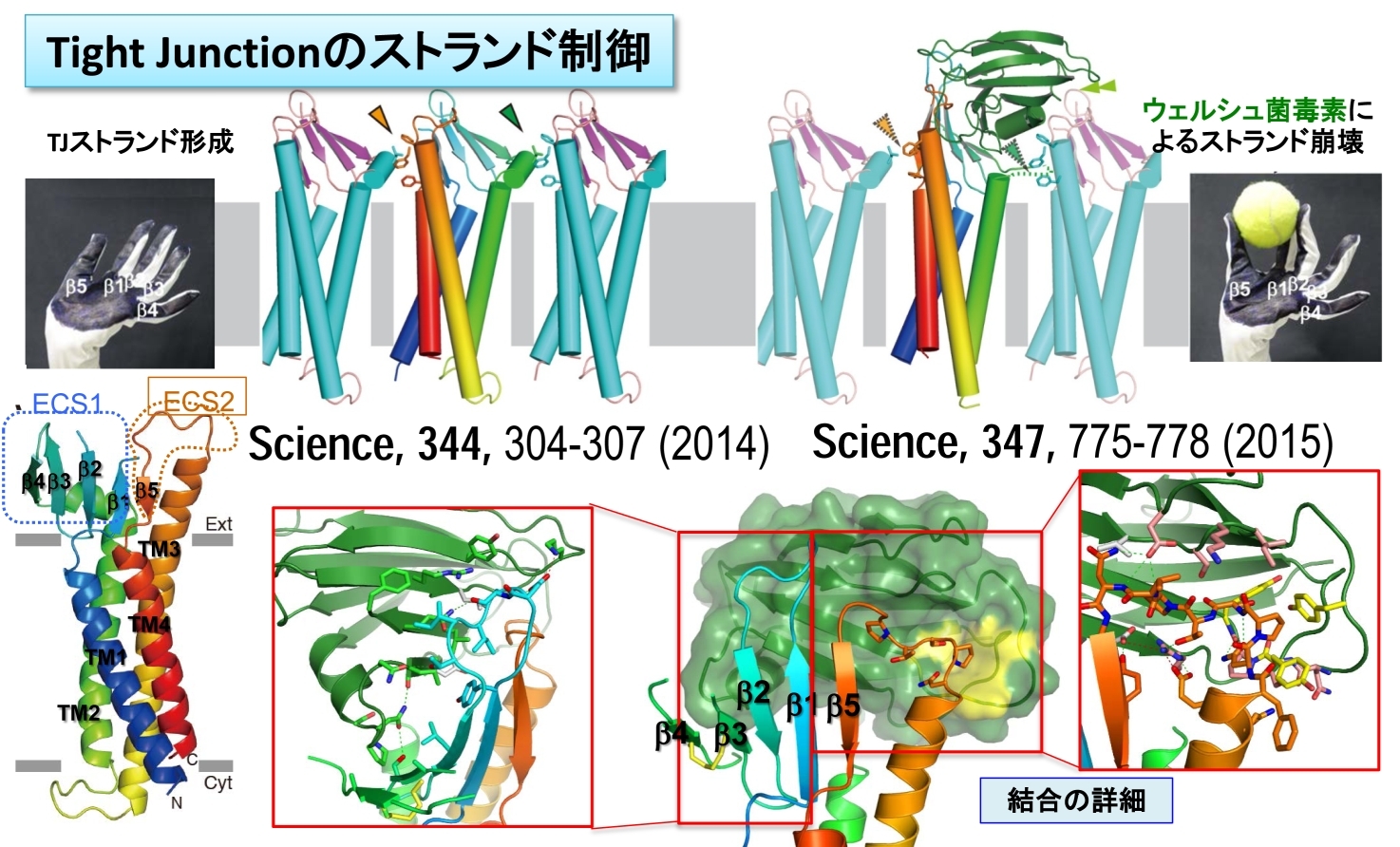
図34 クローディン-19とC-CPEとの構造を解析して提案したTJを崩壊させるモデル。C-CPEが結合していない構造と比較することで、より深く理解できる。
10.4 Cldn19の毒素複合体の構造解析
また、ウェルシュ菌毒素がTJの構造を崩壊させることを確認できたので、TJを制御することで、細胞間の透過制御ができる可能性がある。それゆえ、ウェルシュ菌毒素のC末端側ドメイン(C-CPE)とクローディン-19との複合体の構造を解析して、TJを崩壊させる分子機構を解明した(Science, 347, 775-778, 2015)(図34)。これらの構造情報は、血液脳関門を始めとするパラセルラーチャネルの透過制御薬開発の参考になると考えられるが、血液脳関門をはじめとする細胞間の投下制御は、それほど単純ではないので、より詳細なCldnsの研究の進展が必要不可欠である。
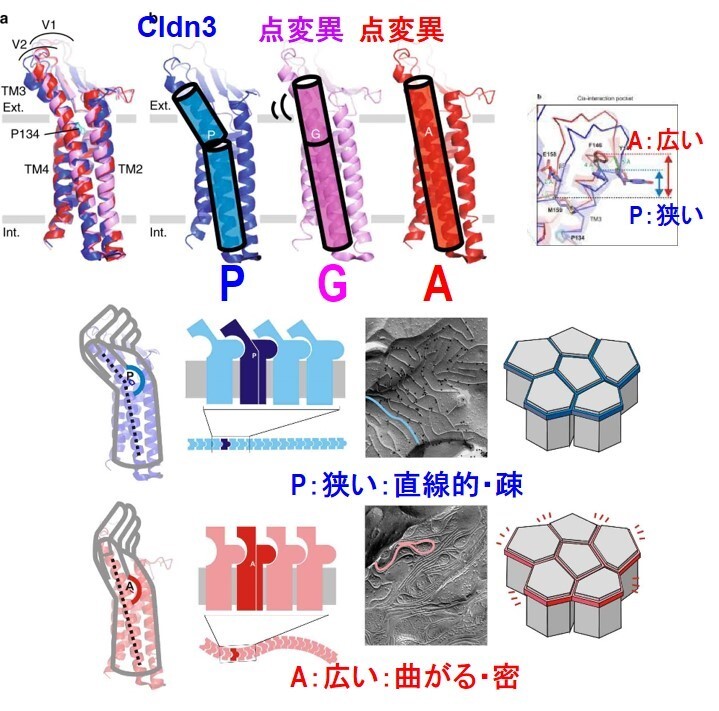
図35 クローディン-3とC-CPEとの複合体の構造に加えて、へリックス3の1残基の変異体の構造を解析して提案したTJストランドの性質を変えるモデル。
10.5 Cldn3と毒素との複合体構造解析
さらにクローディン-3とC-CPEとの複合体の構造を解析するとともに、3番目のヘリックスに存在する134番のプロリンはこのヘリックスの曲がり方に影響を与える。この3番目のヘリックスはCldnがストランドを形成するときの疎水的なポケットの大きさに影響を与える。この3番のヘリックスが曲がると疎水ポケットが狭くなり、ストランドが曲がりにくいが、3番のヘリックスが直線だと疎水ポケットが広くなってストランドは曲がりやすくなる。それゆえ、この132番のアミノ酸をグリシンやアラニンに変異体した場合の構造も解析することによって、クローディンの3番目のヘリックスの傾きに影響する一つのアミノ酸残基の変異によって、TJのストランドの形状が影響を受けることを明らかにした(Nat. Commun., 10, 816, 2019)(図35)。

図36 27種類のクローディン全てをノックアウトした上皮細胞EpH4を作製し、その細胞に1種類だけを発現する27種類の細胞を作製して、それぞれのクローディンの機能を解析した。この膨大な実験によって、クローディンのそれぞれの機能が明らかになり、4つのクラスに分類した。Class 1: 1種類でバリアとなるタイトジャンクションを形成する。Class 2: パラセルラーチャネル機能を有するタイトジャンクションを形成する。Class 3: 自分だけではタイトジャンクションを形成できない。Class 4: 酸性になるとタイトジャンクションのバリア機能を増強する(Cldn18.2)。
タイトジャンクションは上皮細胞の間のアピカル側をシールするバリアを形成しているが、その中心となるクローディンは27種類が知られており、その組み合わせによって、多様な機能を有するタイトジャンクションンが形成されていることがわかっている。しかし、27種類ものクローディン分子それぞれの性質やタイトジャンクションを形成した時の機能は、はっきりしていなかった。共同研究を行っている月田研では、27種類全てのクローディンを欠損させた上皮細胞(EpH4 cell)を作製して、27種類のそれぞれのクローディンの1種類のみを発現して、タイトジャンクションを単独で形成できるか否かと、形成した場合のタイトジャンクションの性質を観察し計測した(Sci. Adv., 11, eadx7431, 2025)。その結果を基に、27種類のクローディンを、大きくは4種類のクラスに分類した(図36)。
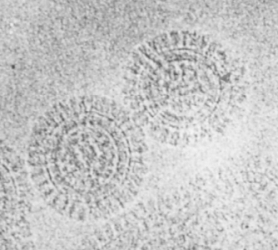
図37 WTのEpH4細胞が形成するタイトジャンクションのクライオ電子線トモグラフィー解析例。
我々は、図33に示すようなタイトジャンクションのanti-parallel double row modelを提案してはいるが、このモデルの正否は確認できていない。それゆえ、タイトジャンクションの構造をクライオ電子線トモグラフィー法で解析して、クローディンがどのようにタイトジャンクションを形成しているか解析できると理想的である。EpH4細胞が形成するタイトジャンクションと、27種類のクローディンを欠損させた細胞間、Cldn2だけ、Cldn3だけで形成されるタイトジャンクションなどの構造を、月田研とDanev研との共同研究で、クライオ電子線トモグラフィー解析した(図37)(Sci. Adv. 11, eadx7431, 2025)。
11.Gタンパク質共役型受容体(X線結晶学)
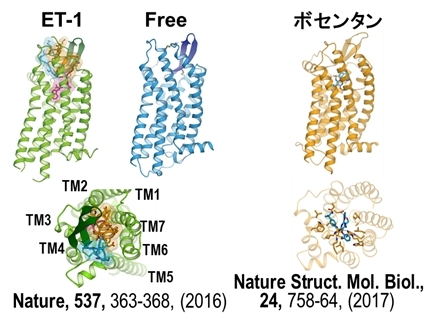
図38 エンドセリンB型受容体とそれにそれぞれのリガンドが結合した構造。
12.単粒子解析法とクライオ電子顕微鏡システム
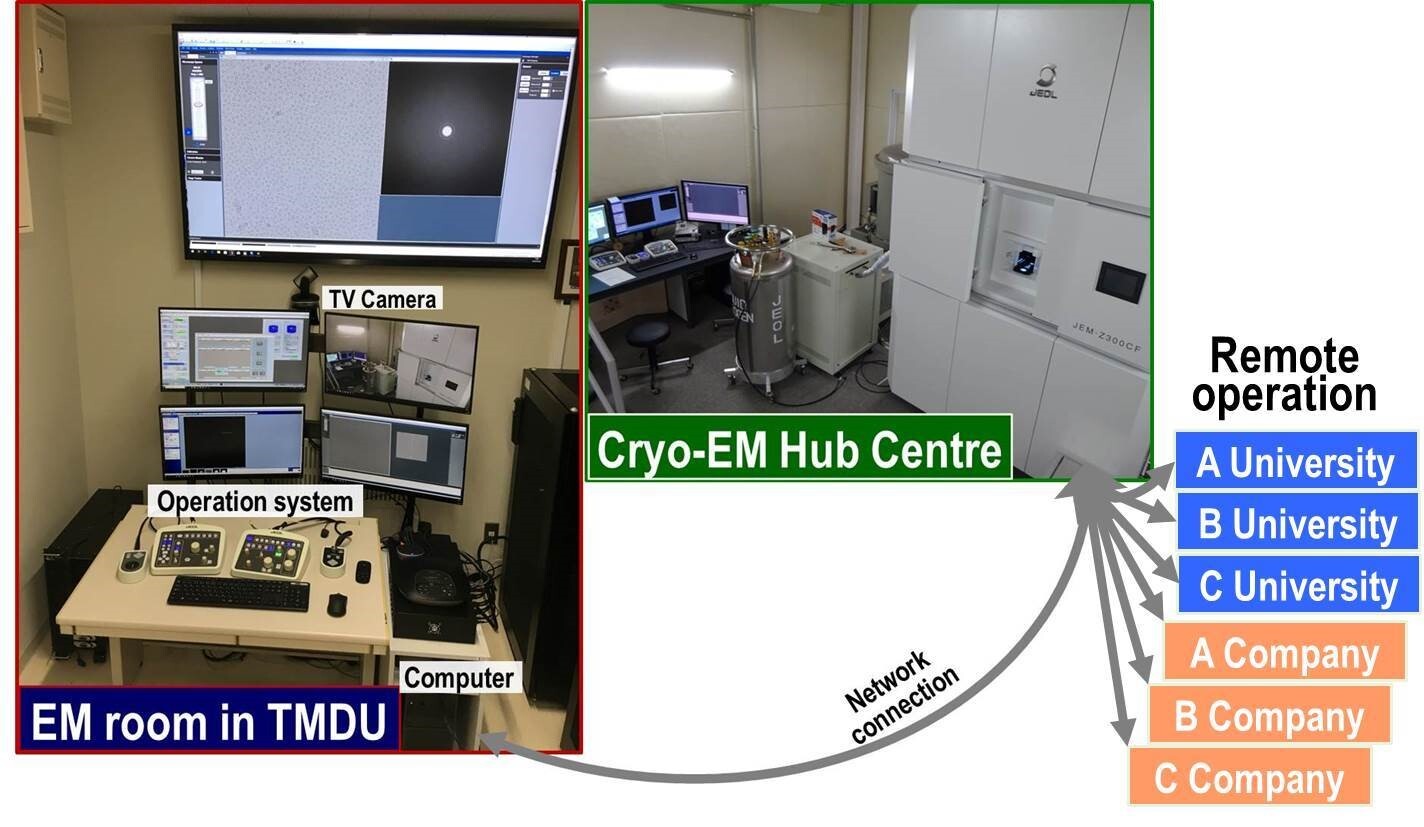
図39 Hub centerに設置したクライオ電子顕微鏡を遠隔操作するシステム。
13.水チャネル(単粒子解析法)
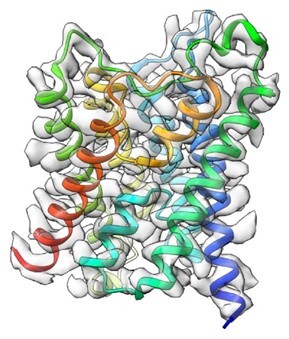
図40 第8世代のクライオ電子顕微鏡を用いて構造解析されたAQP2の構造。
このような単粒子解析が困難な膜タンパク質も、第8世代のクライオ電子顕微鏡を用いて、解析できるようになったので、水チャネルアクアポリン-2(AQP2)の構造解析に成功した(J. Struct. Biol., 215, 107984, 2023)(図40)。
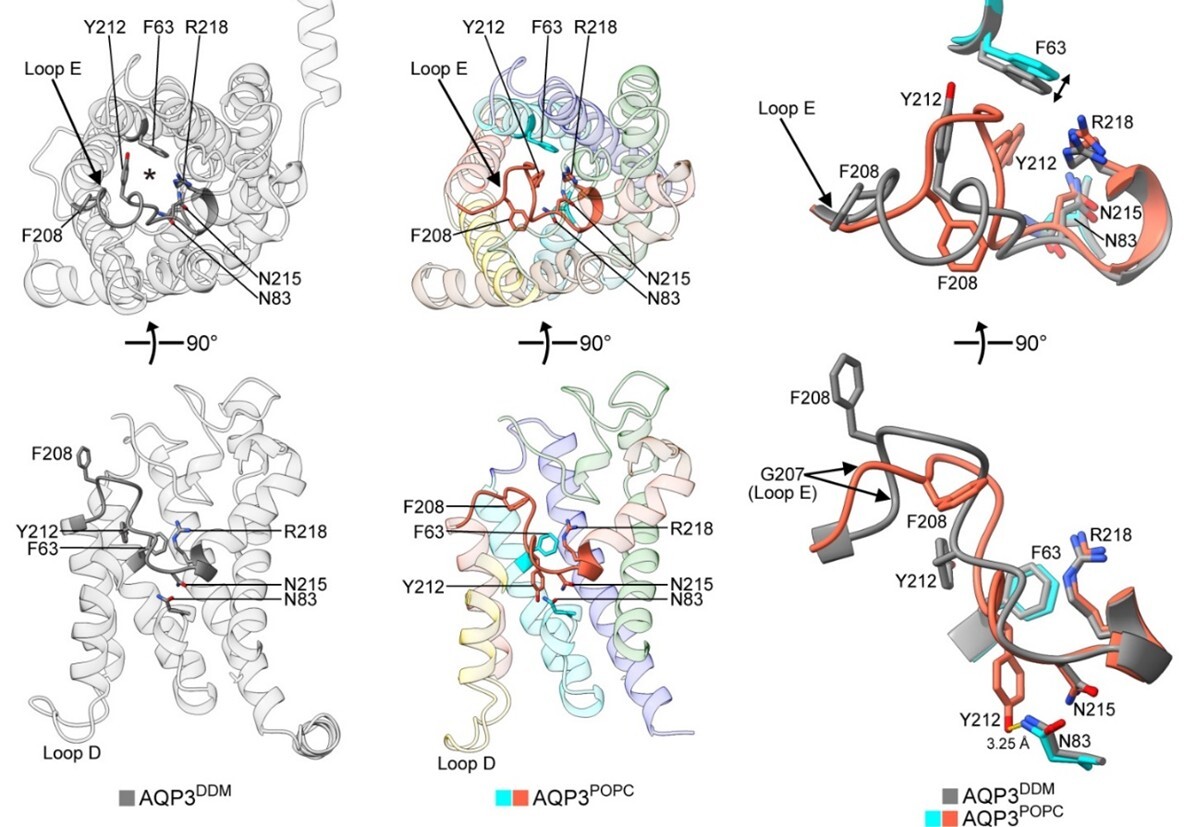
図41 第8世代のクライオ電子顕微鏡を用いて構造解析されたAQP3の構造。DDM中で構造解析されたチャネルが開いた構造(図の左上が細胞外から見た構造で、下が膜面に並行方向から見た4量体を形成するこのチャネルの1つのサブユニットの構造)。POPCで形成されたナノディスク内で解析されたAQP3の構造(図の中上が細胞外から見たチャネルが閉じた構造で、下が膜面に平行方向から見た4量体を形成するこのチャネルの1つのサブユニットで、チャネル内にY212が挿入された構造)。右にチャネルの開閉に関わる部分の構造を重ねて示した構造を示す(図の右上は細胞外から見た構造、右下は膜面に並行方向から見た構造)。グレーが開いた構造、オレンジ色がチャネルが閉じた構造。
13種類の水チャネルの中でも構造解析されていなかったアクアポリン-3(AQP3)の構造解析を行った。その結果驚いたことに、これまでに解析されている水チャネルでは見られなかった構造が明らかになった。すなわち、細胞外側のループ領域に存在するチロシン残基(Y212)がチャネル内に挿入されてチャネルを塞いだ構造をとることが解析された(図41)。それゆえ、AQP3と同じようにグリセロールも透過するaquaglyceroporinである、AQP7とGlpFとの構造を解析したところ、このように細胞外側からループ部分の一部がチャネル内に挿入される構造は見られなかったので、AQP3に特徴的な閉じた構造であると思われる。このAQP3の閉じた構造を形成するには、212番目の位置に存在するアミノ酸が芳香環を持っている必要があることが明らかになった。すなわち、212番目のアミノ酸をフェニルアラニンとスレオニン残基に変異したAQP3の構造を解析したところ、Y212Fの変異AQP3では、変異を導入していないAQP3と同じように、212番のフェニルアラニンがチャネル内に挿入された構造を形成することが明らかになった。一方、Y212Tに変異したAQP3はチャネル内に挿入された構造は見られなかった(Nat. Commun., 16, 2653, 2025)。なお、AQP7とGlpFはAQP3の212に相当する位置に芳香環を有するが、細胞外からチャネルを閉じる構造はとらないことを確認している。
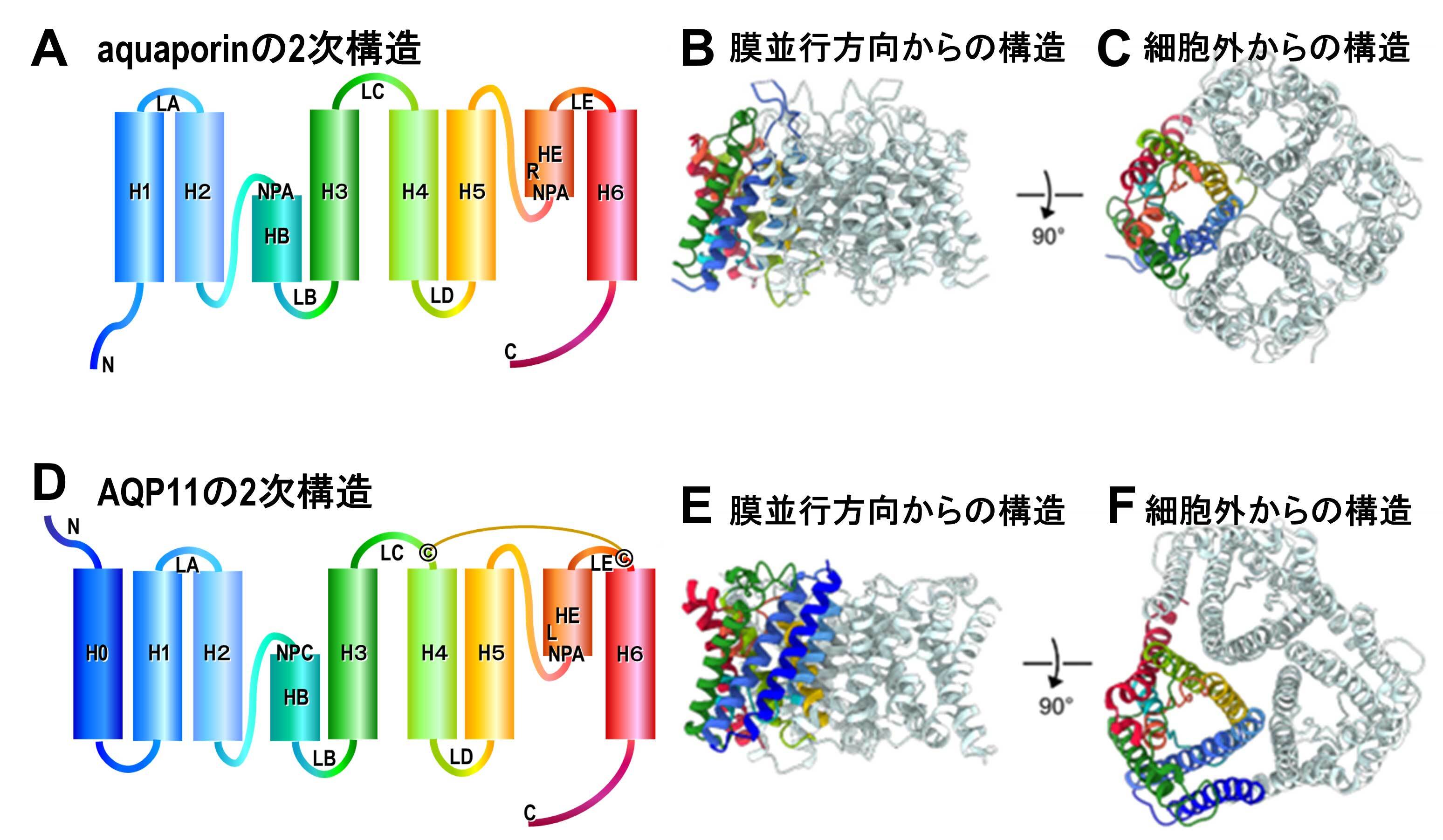
図42 AQP4などの水チャネルの構造(A, B, C)とクライオ電子顕微鏡を用いて単粒子解析したAQP11の構造(D, E, F)比較。AQP11は通常の水チャネルより、N末端側にヘリックス(H0)が形成されており、4量体をとれなくて3量体を形成している。4量体で構造安定化ができない代わりに、LCとLEがS-S結合でつながれて、安定化されている。さらに、ほとんど完全に保存されている、NPAモチーフがLBでNPCとなっている。そして、LEのar/Rと呼ばれるコンストリクションに重要なRがLになっており、AQP11は他の水チャネルより顕著に広いチャネルを形成している。
AQP11は、小胞体に局在した発現がみられるが、そのノックアウトの実験から腎臓が発達する場合に必要不可欠な水チャネルであることが示されている。また、我々は、AQP11が少なくとも水を透過するチャネルであることを示していた(Biochim. Biophys. Acta., 1768, 688-693, 2007, J. Struct. Biol., 174, 315-320, 2011)が、controversialな状況が続いていた。その理由は、水チャネルの機能解析に使われているXenopus oocytesで細胞表面にこのチャネルを発現させて測定することが困難であり、構造が不安定なためであると考えられる。我々は、クライオ電子顕微鏡の単粒子解析法を用いてこのAQP11の構造解析に成功した。その結果は、驚くことに、aquaporinsもaquaglyceroporinsも全て必ずホモ4量体を形成して特徴的なAQP-foldを安定化しているが、AQP11はこのような4量体ではなく、3量体を形成していることが、明らかになった(図42)。すなわち、AQP4などの水チャネルの構造(図42 A, B, C)とクライオ電子顕微鏡を用いて単粒子解析法で解析したAQP11の構造(図42 D, E, F)を比較すると、顕著に異なっている。AQP11は通常の水チャネルより、N末端側に1本ヘリックス(H0)が多く形成されており、4量体をとれなくて3量体を形成している。水チャネルでは、4量体を形成することで構造安定化が行われているが、4量体形成による安定化がないためか、LCとLEがS-S結合でつながれて、構造安定化が図られている。AQP11の重要な特徴として、水チャネルではほとんど完全に保存されている、NPAモチーフがLBではNPCとなっている(図42 D)。そして、ar/Rと呼ばれる狭いチャネルが形成されている部分の重要なアミノ酸残基RがLになっており、AQP11は他の水チャネルより顕著に広いチャネルを形成している。それゆえ、水やグリセロール以外にH2O2などの比較的大きい分子を透過する可能性が示唆されている(Sci. Adv. 12, eaeb5769, 2026)。AQP11をノックアウトすると、腎性嚢胞症などになることと、このチャネルがH2O2などを透過できることは関連しているかもしれない。
14.RNA標的創薬
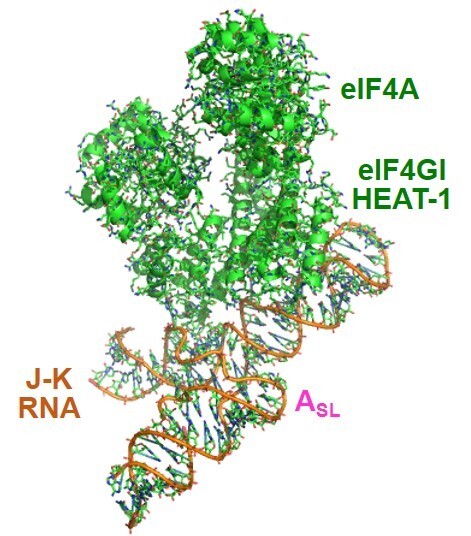
図43 クライオ電子顕微鏡を用いて解析したJ-K-St RNAと宿主細胞の翻訳開始因子との複合体の構造。
15.Gタンパク質共役型受容体(単粒子解析)と構造創薬
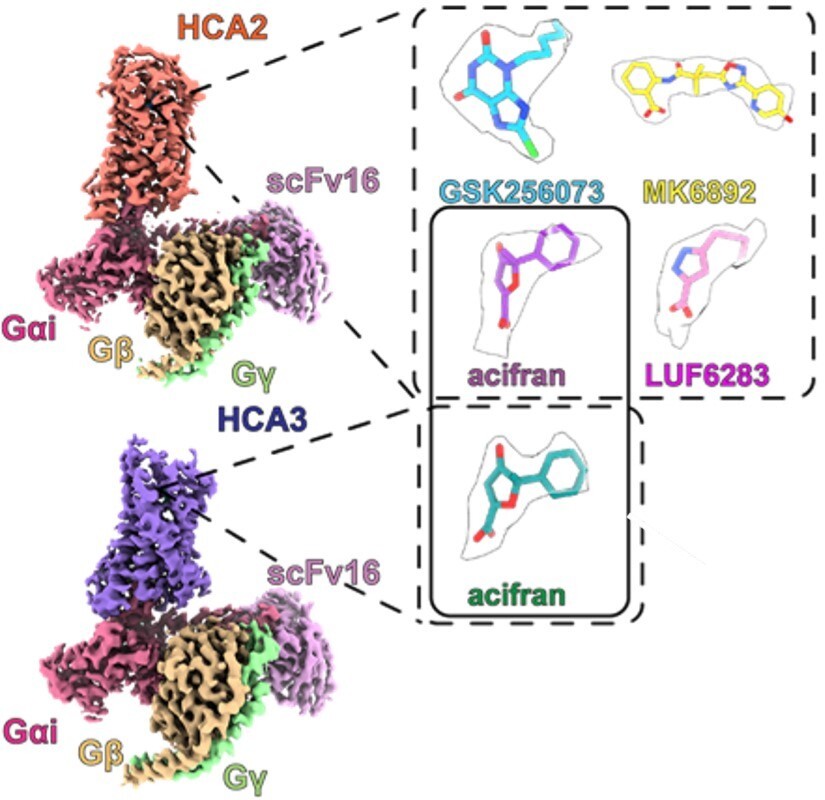
図44 クライオ電子顕微鏡を用いて解析したHCA2とHCA3のリガンド結合構造。
16.構造創薬を活用した新型コロナウイルス治療薬候補の開発
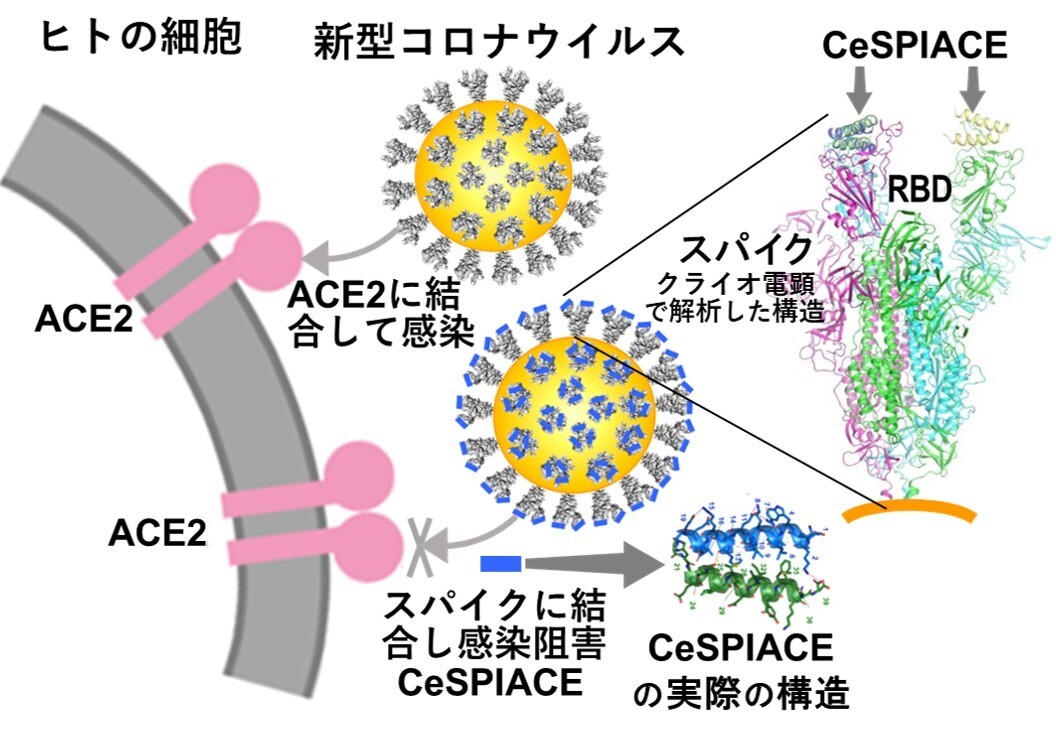
図45 新型コロナウイルスはそのスパイクでACE2に結合することによって人に感染する。それゆえ、スパイクがACE2に結合するために必要不可欠な部分に強く結合して解離しなければ、ウイルスの感染を阻害することができる。スパイクのウイルス外部に突き出したドメイン(エクトドメインと呼ばれる)やACE2に結合するドメインであるRBDの構造をクライオ電子顕微鏡やX線結晶学を用いて高分解能で解析することによって、感染を阻害できる構造形成ペプチドCeSPIACEを設計し、開発することに成功した。スパイク(のRBD)にCeSPIACEが結合した構造も解析した。CeSPIACEは図に示すように2本のヘリックス構造を形成する天然のアミノ酸だけからなる短い構造形成ペプチドで、ウイルスのスパイクだけに結合して他の分子に影響を与えないので、副作用がなく安心して使える強力な治療薬となると期待される。
理想的な薬を開発する創薬戦略として、新型コロナウイルスSARS-CoV-2が、ACE2(Angiotensin-Converting Enzyme 2:アンジオテンシン変換酵素2)に結合するスパイクタンパク質の先端に存在するRBD(Receptor Binding Domain:受容体結合ドメイン)に強く結合する阻害剤を開発することを目指した。しかし、RBDがACE2に結合する接触面には、低分子が結合できるポケットが存在しないため、ペプチド製剤を用いてPPI(Protein-Protein Interaction:タンパク質間相互作用)を制御する必要がある。さらに、新型コロナウイルスなどの変異ウイルスではスパイクタンパク質が繰り返し変異を起こすため、すべての変異ウイルスのスパイクに強く結合できる阻害剤が求められる。そこで、我々は、副作用がなく、安心して使用できる薬の開発を行うために、構造創薬(SGDD: Structure-Guided Drug Development)技術を用いてペプチド医薬候補の開発を試みた。
具体的には、変異を繰り返すウイルスであっても、感染に必要なACE2への結合を阻害することを目指した。RBDがACE2に結合するのと同じRBD部分に強く結合し、解離しない天然のアミノ酸のみで構成される構造形成ペプチドを設計した。開発したペプチドは、武漢株からオミクロン株のXBB.1.5までのすべてのタイプの新型コロナウイルスに強い薬効を示すペプチド医薬候補を開発し、CeSPIACE(COVID-19 eliminative Short-Peptide Inhibiting ACE2 binding)と命名した。
このペプチドは39アミノ酸からなる短い直鎖状分子で、容易に化学合成ができる。同じ長さの2本のヘリックスから成る束を形成し、ヘリックスを巻いた後に塩橋などでその構造を安定化する。また、RBDへの結合面を外側に向けるようにホモ2量体を形成し、4本のヘリックスからなるさらに安定な束の構造を形成するように設計した。この基本構造を保持しながら、ACE2への結合に必要不可欠なRBDのアミノ酸と強く結合できるように設計した(図45)。
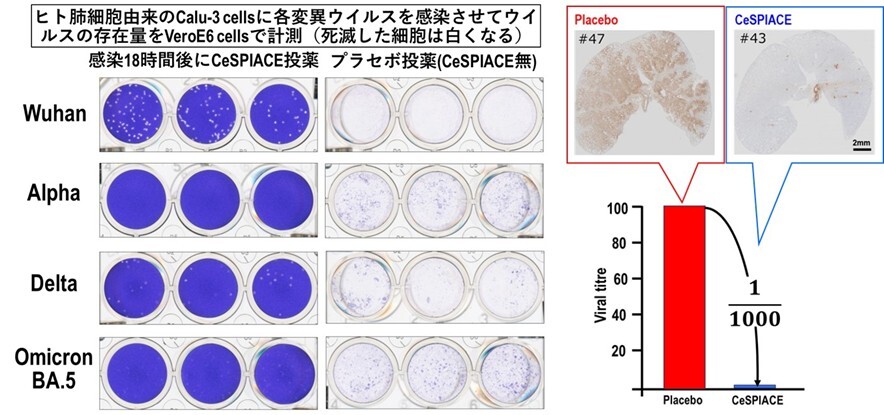
図46 (左図)ヒトの肺細胞由来のCalu-3細胞に、武漢株、アルファ株、デルタ株、オミクロンBA.5株を、それぞれ感染させて、感染18時間後にCeSPIACEを投薬した場合と、CeSPIACEを含まない液を加えた場合の細胞上清をVeroE6/TMPRSS2細胞に加えて、ウイルス量を計測した。細胞がウイルス感染によって死ぬと白くなるので、ウイルス量を観ることができる。CeSPIACEを投薬しないとほとんどの細胞が死滅して白くなっているが、CeSPIACEを加えた場合には、いずれの変異ウイルスに対しても死んだ細胞が激減している。
(右図)デルタ株を感染させたハムスターを用いた実験で、CeSPIACEを含まないプラセボでは肺一面にウイルスが感染しているが、CeSPIACE投与によりウイルスがほとんど見られなくなるほど高い治療効果があることが病理解剖の結果から確認された(右上)。ウイルスRNAを測定した結果、1/1000にウイルス量が減っていることが確認された(右下)。
実験では、武漢型、アルファ型、デルタ型、オミクロン株(BA.1、BA.2、BA.5)のRBDに対するCeSPIACEの解離定数(KD値)を測定した結果、1nM以下の強い結合能を確認した。また、感染細胞を用いた抗ウイルス活性の半数阻害濃度(IC50)は10nM以下で、強い抗ウイルス活性を確認した。さらに、ヒト肺細胞由来のCalu-3細胞を用いた実験でも、多様な変異ウイルスに対して強い抗ウイルス活性を確認した(図46左)。加えて、シリアンハムスターを用いた動物実験では、プラセボ条件と比較してウイルスRNA量を1/1000に減少させる顕著な抗ウイルス活性を確認した(図46右)。
新型コロナウイルスの感染状況や、ワクチン・治療薬開発の状況を踏まえ、ウイルス感染を強力に阻害し、副作用がなく安全で使いやすい薬の必要性を感じたことから、本研究の開発を開始した(2020年の6月から開発を開始した)ために、パンデミックに間に合う形で治療に使えるようにはできなかった。しかし、当初の創薬戦略で掲げた「副作用がなく、躊躇なく安心して使用できる薬」を開発するという目標に基づき、候補物質としてペプチド医薬CeSPIACEを開発することに成功した。この構造形成ペプチドCeSPIACEは、ウイルスが変異を繰り返しても、ヒトのACE2に結合して感染する限り全てのウイルススパイクに対応可能と期待できる。天然のアミノ酸のみで構成されているため、副作用の懸念がなく、軽症から重症までの感染者に対しても強い薬効が期待される(Proc. Natl. Acad. Sci. U. S. A., 122, e2413465122, 2025)。
さらに、CeSPIACEは治療薬としてだけでなく予防薬としての利用も可能であり、抗体薬に比べて安価に大量供給が可能である。これは、化学合成が容易な直鎖状の短い構造形成ペプチドであることに加え、構造創薬技術を活用することで短期間での開発と改良が可能になったことによって開発できた。また、室温での高い安定性と優れた水溶性を備えた物性により、品質管理が容易で、多くの国や地域に供給可能と考えられる。それゆえ、このような特性を持つCeSPIACEは、将来のパンデミックへの備えとして大きな社会的意義を持つだけでなく、世界中の感染症治療に新たな希望を提供する可能性を秘めている。
なお、以上の研究成果は、本文中に記載した共同研究者だけでなく、それぞれの論文の著者諸氏との共同研究によるものであり、共同研究者の皆様に感謝する。
| Drug Rescuing: 製薬企業などには、創薬標的として有望な分子とそのリード化合物が得られていても、前臨床、臨床などの過程において問題が生じたために薬が販売できなかった例が、多く蓄積されていると思われる。これらの創薬標的とリガンドとの複合体の構造を解析することによってリガンド結合の詳細な構造情報が得られるので、標的分子とリガンドとの相互作用を再設計することが出来る。また、リガンド結合に影響を与えないリガンド部分を知ることが出来る。この部分の化学構造を結合能に影響を与えることなく改変することによって、副作用が軽減できる可能性がある。この様に、詳細な構造情報に基づいて、薬となりきれていなかった創薬標的とそれと結合する候補化合物を薬として作りきる効率の良い創薬戦略をこの様に命名している。 |