
「新規オートファジーの変調は神経変性疾患の原因となる」【清水重臣 教授、荒川聡子講師、山口啓史助教】

清水 重臣 (シミズ シゲオミ)教授 難治疾患研究所 病態細胞生物学分野(中央)
荒川 聡子 (アラカワ サトコ)講師 同上(右)
山口 啓史 (ヤマグチ ヒロフミ)助教 同上(左)
| ● | オートファジー*1は細胞構成成分を分解する機構で、その異常は多くの疾患の発症に関わります。 |
| ● | 研究グループは以前の研究で、Atg5, Atg7等に依存しない第二のオートファジー機構を発見しました。 |
| ● | この新規オートファジーを制御する鍵分子は不明でしたが、今回実行分子の同定に成功しました。 |
| ● | この実行分子を脳特異的に欠損させたマウスでは、神経変性が生じました。 |
| ● | 従来型オートファジーと新規オートファジーは、各々異なるメカニズムで神経細胞を守っていることがわかりました。 |
| ● | 今回の研究成果は神経変性疾患の新規治療法開発に新たなヒントを与えるものです。 |
東京医科歯科大学・難治疾患研究所・病態細胞生物分野の荒川聡子講師、清水重臣教授らの研究グループは、産業技術総合研究所、東京大学、日本医科大学との共同研究で、細胞内のタンパク質を分解する新しい仕組みを実行する分子を発見しました。この分子は神経変性疾患に関与していると考えられ、将来的に新規治療法開発への応用が期待されます。この研究は文部科学省科学研究費補助金、日本医療研究開発機構などの支援のもとでおこなわれたもので、その研究成果は、国際科学誌Nature Communicationsに、2020年10月20日午前10時(英国夏時間)にオンライン版で発表されました。
研究の背景
オートファジーは、細胞内構成成分の分解機構です。栄養飢餓時に誘導されるオートファジーの分子機構は良く解析されており、必須のタンパク質であるAtg5, Atg7, LC3*2等によって調節され、小胞体膜を起源とすることが明らかになっています。一方で、研究グループは、この従来型オートファジーとは異なり、Atg5などを使わず、ゴルジ体膜を起源とする第2のオートファジー機構を見出していました(新規オートファジー、図1上、Nature 2009年)。今回の研究は、この新規オートファジーのメカニズムならびに生体での役割を明らかにした研究です。
研究成果の概要
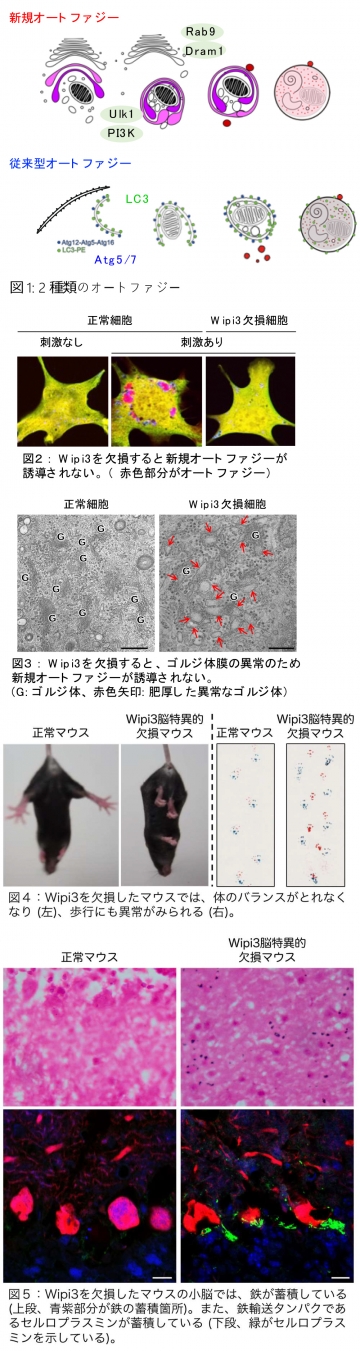
研究グループは、新しいタンパク質分解機構である新規オートファジーを2009年に発見しました。その後の研究で、その新規オートファジーが、酵母から哺乳動物まで保存されていることがわかりました。
今回の研究では、はじめに酵母を用いて実験を行い、この新規オートファジーに関わるHsv2という遺伝子を見つけました。そこで、酵母のHsv2の哺乳動物相同遺伝子であるWipi3を欠損させた細胞を作製し、新規オートファジー誘導刺激を加えました。すると、正常細胞では観察される新規オートファジー(図2:中央の細胞、赤色部分がオートファジー)が、Wipi3欠損細胞では観察されませんでした(図2:右の細胞)。
この新規オートファジーは、従来のオートファジーと異なり、ゴルジ体膜からオートファジーが形成されることがわかっています(従来型のオートファジーは、ゴルジ体ではなく、小胞体膜からできます)。そこで、Wipi3欠損細胞を電子顕微鏡で観察したところ、ゴルジ体膜の形態が異常であり、ゴルジ体膜の異常のため新規オートファジーが誘導されないことがわかりました(図3)。
次に、Wipi3の細胞内局在がどこにあるのかについて、最新のイメージング技術である超解像顕微鏡を使って観察しました。その結果、細胞質に散在していたWipi3が、新規オートファジーの刺激を受けると、ゴルジ体の膜上に移動することがわかりました。即ち、新規オートファジーが起きるときは、Wipi3が細胞質からゴルジ体に移動し、ゴルジ体の膜を変形させて、オートファジーの膜をつくっていることがわかりました。
最後に、神経細胞でWipi3遺伝子をもたないマウスを作製しました。すると、Wipi3欠損マウスでは、体のバランスをとれなくなり、歩行にも異常がみられました(図4)。そこで、動物の行動を制御する小脳を調べたところ、(1)小脳のプルキンエ細胞が変性脱落していること、(2)脱落前の神経細胞でゴルジ体の形態が異常であること、(3)新規オートファジーが起きていないこと、(4)鉄ならびに鉄輸送タンパク質であるセルロプラスミンが蓄積していること(図5)、(5)セルロプラスミンは新規オートファジーで分解されるタンパク質であるため、この分解不全によって鉄沈着が認められること、がわかりました。
従来型オートファジーに必須のAtg7を欠損したマウスの脳でも同様の神経機能異常が見られますが、両者の神経細胞の内部構造は全く異なっており、各々異なる機構で神経機能を維持していることが考えられました。実際に、Wipi3, Atg7の両者を欠損したマウスを作成したところ、生後28日で死亡し、単独の遺伝子欠損マウスよりもはるかに重篤な神経変性を示すことがわかりました。このことから、従来型オートファジーと新規オートファジーは、異なる機構で神経細胞を維持していることが確認されました。
なお、ヒトの脳内鉄沈着神経変性症SENDA*3は、Wipi4(Wipi3の相同遺伝子)の遺伝子変異によって発症することが知られていますが、Wipi4欠損マウスでは、脳内鉄沈着が見られず症状もあまり見られないのに対して、Wipi3欠損マウスでは、脳内鉄沈着が認められ症状も強いため、SENDAのモデルマウスと考えられます。
今回の研究では、はじめに酵母を用いて実験を行い、この新規オートファジーに関わるHsv2という遺伝子を見つけました。そこで、酵母のHsv2の哺乳動物相同遺伝子であるWipi3を欠損させた細胞を作製し、新規オートファジー誘導刺激を加えました。すると、正常細胞では観察される新規オートファジー(図2:中央の細胞、赤色部分がオートファジー)が、Wipi3欠損細胞では観察されませんでした(図2:右の細胞)。
この新規オートファジーは、従来のオートファジーと異なり、ゴルジ体膜からオートファジーが形成されることがわかっています(従来型のオートファジーは、ゴルジ体ではなく、小胞体膜からできます)。そこで、Wipi3欠損細胞を電子顕微鏡で観察したところ、ゴルジ体膜の形態が異常であり、ゴルジ体膜の異常のため新規オートファジーが誘導されないことがわかりました(図3)。
次に、Wipi3の細胞内局在がどこにあるのかについて、最新のイメージング技術である超解像顕微鏡を使って観察しました。その結果、細胞質に散在していたWipi3が、新規オートファジーの刺激を受けると、ゴルジ体の膜上に移動することがわかりました。即ち、新規オートファジーが起きるときは、Wipi3が細胞質からゴルジ体に移動し、ゴルジ体の膜を変形させて、オートファジーの膜をつくっていることがわかりました。
最後に、神経細胞でWipi3遺伝子をもたないマウスを作製しました。すると、Wipi3欠損マウスでは、体のバランスをとれなくなり、歩行にも異常がみられました(図4)。そこで、動物の行動を制御する小脳を調べたところ、(1)小脳のプルキンエ細胞が変性脱落していること、(2)脱落前の神経細胞でゴルジ体の形態が異常であること、(3)新規オートファジーが起きていないこと、(4)鉄ならびに鉄輸送タンパク質であるセルロプラスミンが蓄積していること(図5)、(5)セルロプラスミンは新規オートファジーで分解されるタンパク質であるため、この分解不全によって鉄沈着が認められること、がわかりました。
従来型オートファジーに必須のAtg7を欠損したマウスの脳でも同様の神経機能異常が見られますが、両者の神経細胞の内部構造は全く異なっており、各々異なる機構で神経機能を維持していることが考えられました。実際に、Wipi3, Atg7の両者を欠損したマウスを作成したところ、生後28日で死亡し、単独の遺伝子欠損マウスよりもはるかに重篤な神経変性を示すことがわかりました。このことから、従来型オートファジーと新規オートファジーは、異なる機構で神経細胞を維持していることが確認されました。
なお、ヒトの脳内鉄沈着神経変性症SENDA*3は、Wipi4(Wipi3の相同遺伝子)の遺伝子変異によって発症することが知られていますが、Wipi4欠損マウスでは、脳内鉄沈着が見られず症状もあまり見られないのに対して、Wipi3欠損マウスでは、脳内鉄沈着が認められ症状も強いため、SENDAのモデルマウスと考えられます。
研究成果の意義
今回の研究成果では、新規オートファジーにおいて、Wipi3が重要な実行分子であることを発見しました。また、新規オートファジーは神経細胞の維持に必要不可欠であることを見出しました。さらに、新規オートファジーの変調は、ヒトの脳内鉄沈着神経変性症SENDAの発症原因となりうる可能性が示されました。今回の研究成果は、神経変性疾患の病態生理解明や疾患治療法開発に波及効果をもたらすと期待できます。
用語解説
※1オートファジー
細胞内のタンパク質や細胞内小器官を分解する細胞機能。オートファジーではまず細胞内のタンパク質や細胞内小器官が隔離膜によって取り囲まれてオートファゴソームが形成される。次にオートファゴソームの外膜がリソソーム膜と融合しオートリソソームとなり、内容物が分解される。この細胞機能が破綻すると、神経変性疾患を含めた様々な疾患の発症に繋がることがわかっている。
※2Atg5, Atg7, LC3
LC3は酵母Atg8の哺乳類でのタンパク質の一つ。これらはオートファジーを進行するために必須なタンパク質である。
※3SENDA
脳内鉄沈着神経変性症の一つであり、小児期早期からの非進行性の知的障害と、成人期に急速に進行する錐体外路症状(ジストニアやパーキンソン様症状)、認知症を呈する神経変性疾患である。
細胞内のタンパク質や細胞内小器官を分解する細胞機能。オートファジーではまず細胞内のタンパク質や細胞内小器官が隔離膜によって取り囲まれてオートファゴソームが形成される。次にオートファゴソームの外膜がリソソーム膜と融合しオートリソソームとなり、内容物が分解される。この細胞機能が破綻すると、神経変性疾患を含めた様々な疾患の発症に繋がることがわかっている。
※2Atg5, Atg7, LC3
LC3は酵母Atg8の哺乳類でのタンパク質の一つ。これらはオートファジーを進行するために必須なタンパク質である。
※3SENDA
脳内鉄沈着神経変性症の一つであり、小児期早期からの非進行性の知的障害と、成人期に急速に進行する錐体外路症状(ジストニアやパーキンソン様症状)、認知症を呈する神経変性疾患である。
論文情報
掲載誌: Nature Communications
論文タイトル:Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration
論文タイトル:Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration
研究者プロフィール
清水 重臣 (シミズ シゲオミ) Shimizu Shigeomi
東京医科歯科大学
難治疾患研究所 病態細胞生物学分野 教授
・研究領域
オートファジー
細胞死
オルガネラ・バイオロジー
荒川 聡子 (アラカワ サトコ) Arakawa Satoko
東京医科歯科大学
難治疾患研究所 病態細胞生物学分野 講師
・研究領域
オートファジー
細胞死
微細構造生物学
山口 啓史 (ヤマグチ ヒロフミ) Hirofumi Yamaguchi
東京医科歯科大学
難治疾患研究所 病態細胞生物学分野 助教
・研究領域
オートファジー
細胞生物学
東京医科歯科大学
難治疾患研究所 病態細胞生物学分野 教授
・研究領域
オートファジー
細胞死
オルガネラ・バイオロジー
荒川 聡子 (アラカワ サトコ) Arakawa Satoko
東京医科歯科大学
難治疾患研究所 病態細胞生物学分野 講師
・研究領域
オートファジー
細胞死
微細構造生物学
山口 啓史 (ヤマグチ ヒロフミ) Hirofumi Yamaguchi
東京医科歯科大学
難治疾患研究所 病態細胞生物学分野 助教
・研究領域
オートファジー
細胞生物学
問い合わせ先
<研究に関すること>
東京医科歯科大学大学院医歯学総合研究科
病態細胞生物学分野 清水 重臣(シミズシゲオミ)
TEL:03-5803-4692 FAX:03-5803-4821
E-mail:shimizu.pcb@mri.tmd.ac.jp
東京医科歯科大学大学院医歯学総合研究科
病態細胞生物学分野 荒川 聡子(アラカワサトコ)
TEL:03-5803-4797 FAX:03-5803-4821
E-mail:arako.pcb@mri.tmd.ac.jp
<報道に関すること>
東京医科歯科大学 総務部総務秘書課広報係
〒113-8510 東京都文京区湯島1-5-45
TEL:03-5803-5833 FAX:03-5803-0272
E-mail:kouhou.adm@tmd.ac.jp
東京医科歯科大学大学院医歯学総合研究科
病態細胞生物学分野 清水 重臣(シミズシゲオミ)
TEL:03-5803-4692 FAX:03-5803-4821
E-mail:shimizu.pcb@mri.tmd.ac.jp
東京医科歯科大学大学院医歯学総合研究科
病態細胞生物学分野 荒川 聡子(アラカワサトコ)
TEL:03-5803-4797 FAX:03-5803-4821
E-mail:arako.pcb@mri.tmd.ac.jp
<報道に関すること>
東京医科歯科大学 総務部総務秘書課広報係
〒113-8510 東京都文京区湯島1-5-45
TEL:03-5803-5833 FAX:03-5803-0272
E-mail:kouhou.adm@tmd.ac.jp