
研究内容
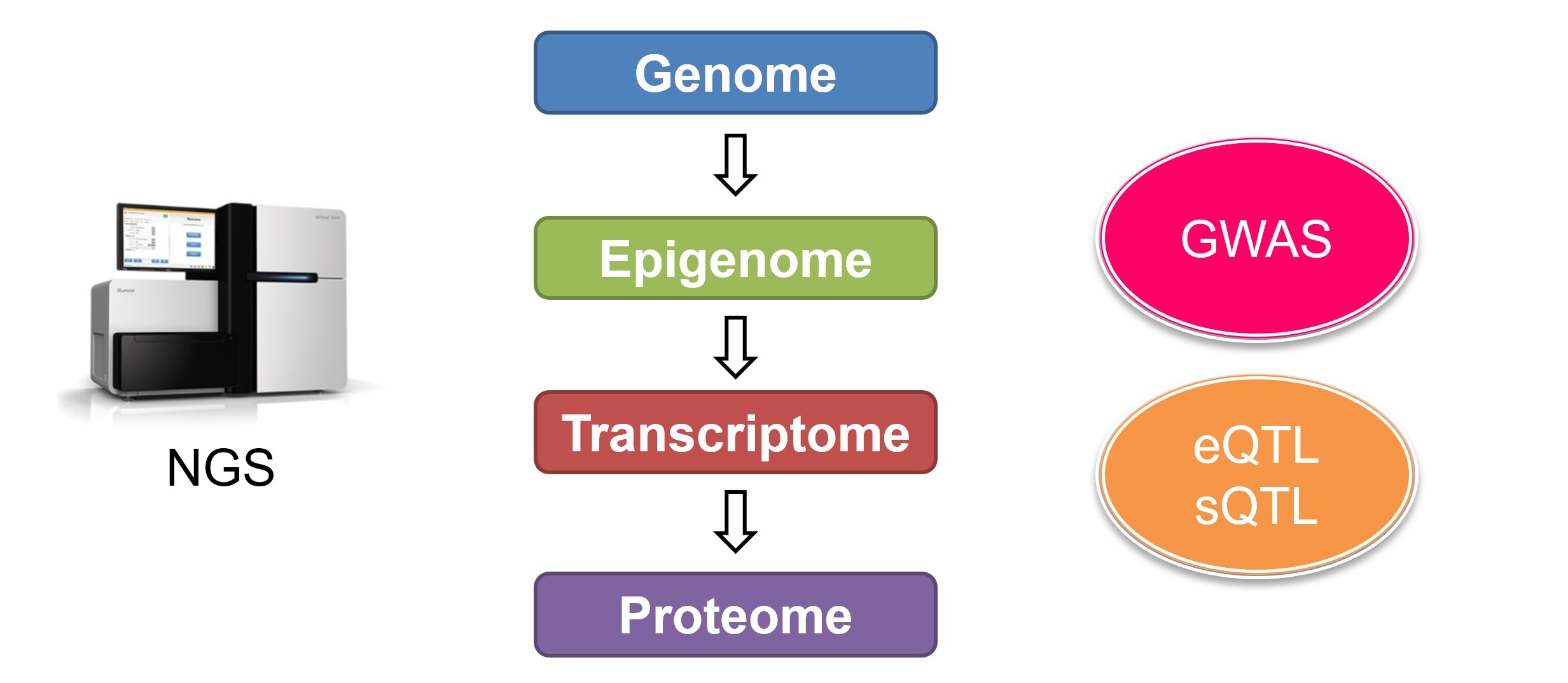
本分野では、ポストGWAS(Genome-Wide Association Study)研究として、エピゲノムやトランスクリプトームなどの多様なビッグデータを最新のAI技術(ゲノム基盤モデル)で解析し、さらに分子生物学的手法を組み合わせることで、多因子疾患の病態解明に向けた多面的なアプローチを試みます。さらに得られた知見に基づき、個人のゲノム情報から病態や薬剤応答性を予測する手法を開発し、プレシジョン医療の確立を目指します。
1)遺伝子多型の網羅的機能解析
多因子疾患のリスクとなる遺伝子多型の多くは、遺伝子発現に影響を及ぼす領域、すなわち eQTL(expression quantitative trait locus)であると考えられています。GTEx Project をはじめとする大規模 eQTL 解析データが公開されており、我々も日本人の免疫細胞サブセットにおける QTL カタログの構築を進めてきました(Nat Genet 2017)。
また、遺伝子多型がスプライシングに影響を与える領域、すなわち sQTL(splicing QTL)も疾患機序において重要な役割を果たすと考えられています。研究室では、従来のショートリードシークエンサーによる eQTL データに加え、ロングリードシークエンサー(後述)を用いたスプライスアイソフォーム解析を組み合わせることで、疾患に関わる sQTL の全貌解明を目指しています。
さらに近年、AlphaGenome や Evo2 などの AI 技術を用いたゲノム基盤モデルが登場し、in silico による遺伝子多型の機能予測が現実味を帯びつつあります。これらの最先端の解析技術を用いることで、疾患の原因となっている遺伝子多型の機能解析を行います。
2)多因子疾患の原因遺伝子機能解析
GWASは、疾患毎に100を超える感受性遺伝子領域を明らかにしてきました。一つ一つの遺伝因子の果たす役割は小さいですが、病態の一側面を形成しているのも事実です。したがって、個々の感受性遺伝子の機能を明らかにすることが病態解明の第一歩となります。たとえば、我々は筋無症候性皮膚筋炎(CADM)のGWASにおいて、WDFY4遺伝子のsQTL効果を持つ多型が疾患と関連することを明らかにしましたが、リスクアレルによって増加するC末端欠如型のWDFY4タンパクが、RNAウィルス認識受容体であるMDA5のシグナルを増強することを明らかにしました(Ann Rheum Dis 2018)。CADMは、致死率が50%にいたる急速進行性の間質性肺炎をもたらすため、このC末端欠如型WDFY4タンパクを標的とした治療法が有望であると考えられます。このようにGWAS候補領域の詳細な機能解析によって疾患の治療に直結する知見を得られる可能性があります。
3)システムズ・アプローチによる疾患の病態予測
個々の遺伝因子を解析することによって、多因子疾患の病態の一側面が明らかになりますが、病態全体を形成するのはこれらの遺伝因子の積み重なりです。したがって、疾患をシステムとみなしたうえで、この遺伝因子の積み重なりをシステムズ・アプローチによって解析することが、病態の全体像や個人間の病態の違いを評価するために必要です。実際に我々は、CD4陽性T細胞におけるeQTLの積み重なり(polygenic score)がTNF-αの活性化に寄与していることを、関節リウマチ患者のゲノムを評価することによって明らかにしました(Nat Genet 2017)。当分野では、GWAS、eQTL、sQTL、エピゲノムなどの様々なオミックスデータを統合することによって、ゲノム情報を用いた疾患の病態予測法の樹立を目指します。また、バイオバンクジャパンやUK BioBankなどの大規模ゲノムコホートデータを用いて予測モデルの検証を行い、プレシジョン医療の実現化を目指します。
4) ロングリード・シークエンサー用いた疾患オミックス解析
現在、第4世代シークエンス技術であるロングリード・シークエンサーが、疾患オミックス解析に革新をもたらしつつあります。たとえば、Nanopore社のシークエンサーは、DNAやRNAがポアを通過するときに発生する電流変化から、1万塩基以上を連続して読むことが可能であり、リピート配列や構造多型の解析に適しています。また、RNA解析では、様々なスプライス・アイソフォームの同定に加えて、リピート配列由来転写産物の同定も可能です。本分野では、ロングリード・シークエンサーを用いることによって、希少遺伝性疾患、神経疾患、癌などの病態・病因解明を目指しています。さらに、現在知られているタンパク質に翻訳される領域は、ヒトゲノムの数%しか占めていません。その他の部分の多くは、レトロウィルス由来の配列であるレトロトランスポゾンなどの反復配列が占めています。これらの反復配列の中には、未知の翻訳される配列が存在している可能性があります。ロングリード・シークエンサーや分子生物学的な手法を用いて、反復配列由来の新規な転写産物の機能についても解析しています。
1. Ishigaki K, Kochi Y et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat Genet 49:1120-1125,2017
2. Kochi Y, Kamatani Y et al. Splicing variant of WDFY4 augments MDA5 signalling and the risk of clinically amyopathic dermatomyositis. Ann Rheum Dis 77:602-611,2018
3. Sone J, Mitsuhashi S et al Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 51:1215-1221, 20191)
4. Honda S, Kochi Y et al. Association of Polygenic Risk Scores With Radiographic Progression in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2022, AOP.
1)遺伝子多型の網羅的機能解析
多因子疾患のリスクとなる遺伝子多型の多くは、遺伝子発現に影響を及ぼす領域、すなわち eQTL(expression quantitative trait locus)であると考えられています。GTEx Project をはじめとする大規模 eQTL 解析データが公開されており、我々も日本人の免疫細胞サブセットにおける QTL カタログの構築を進めてきました(Nat Genet 2017)。
また、遺伝子多型がスプライシングに影響を与える領域、すなわち sQTL(splicing QTL)も疾患機序において重要な役割を果たすと考えられています。研究室では、従来のショートリードシークエンサーによる eQTL データに加え、ロングリードシークエンサー(後述)を用いたスプライスアイソフォーム解析を組み合わせることで、疾患に関わる sQTL の全貌解明を目指しています。
さらに近年、AlphaGenome や Evo2 などの AI 技術を用いたゲノム基盤モデルが登場し、in silico による遺伝子多型の機能予測が現実味を帯びつつあります。これらの最先端の解析技術を用いることで、疾患の原因となっている遺伝子多型の機能解析を行います。
2)多因子疾患の原因遺伝子機能解析
GWASは、疾患毎に100を超える感受性遺伝子領域を明らかにしてきました。一つ一つの遺伝因子の果たす役割は小さいですが、病態の一側面を形成しているのも事実です。したがって、個々の感受性遺伝子の機能を明らかにすることが病態解明の第一歩となります。たとえば、我々は筋無症候性皮膚筋炎(CADM)のGWASにおいて、WDFY4遺伝子のsQTL効果を持つ多型が疾患と関連することを明らかにしましたが、リスクアレルによって増加するC末端欠如型のWDFY4タンパクが、RNAウィルス認識受容体であるMDA5のシグナルを増強することを明らかにしました(Ann Rheum Dis 2018)。CADMは、致死率が50%にいたる急速進行性の間質性肺炎をもたらすため、このC末端欠如型WDFY4タンパクを標的とした治療法が有望であると考えられます。このようにGWAS候補領域の詳細な機能解析によって疾患の治療に直結する知見を得られる可能性があります。
3)システムズ・アプローチによる疾患の病態予測
個々の遺伝因子を解析することによって、多因子疾患の病態の一側面が明らかになりますが、病態全体を形成するのはこれらの遺伝因子の積み重なりです。したがって、疾患をシステムとみなしたうえで、この遺伝因子の積み重なりをシステムズ・アプローチによって解析することが、病態の全体像や個人間の病態の違いを評価するために必要です。実際に我々は、CD4陽性T細胞におけるeQTLの積み重なり(polygenic score)がTNF-αの活性化に寄与していることを、関節リウマチ患者のゲノムを評価することによって明らかにしました(Nat Genet 2017)。当分野では、GWAS、eQTL、sQTL、エピゲノムなどの様々なオミックスデータを統合することによって、ゲノム情報を用いた疾患の病態予測法の樹立を目指します。また、バイオバンクジャパンやUK BioBankなどの大規模ゲノムコホートデータを用いて予測モデルの検証を行い、プレシジョン医療の実現化を目指します。
4) ロングリード・シークエンサー用いた疾患オミックス解析
現在、第4世代シークエンス技術であるロングリード・シークエンサーが、疾患オミックス解析に革新をもたらしつつあります。たとえば、Nanopore社のシークエンサーは、DNAやRNAがポアを通過するときに発生する電流変化から、1万塩基以上を連続して読むことが可能であり、リピート配列や構造多型の解析に適しています。また、RNA解析では、様々なスプライス・アイソフォームの同定に加えて、リピート配列由来転写産物の同定も可能です。本分野では、ロングリード・シークエンサーを用いることによって、希少遺伝性疾患、神経疾患、癌などの病態・病因解明を目指しています。さらに、現在知られているタンパク質に翻訳される領域は、ヒトゲノムの数%しか占めていません。その他の部分の多くは、レトロウィルス由来の配列であるレトロトランスポゾンなどの反復配列が占めています。これらの反復配列の中には、未知の翻訳される配列が存在している可能性があります。ロングリード・シークエンサーや分子生物学的な手法を用いて、反復配列由来の新規な転写産物の機能についても解析しています。
1. Ishigaki K, Kochi Y et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat Genet 49:1120-1125,2017
2. Kochi Y, Kamatani Y et al. Splicing variant of WDFY4 augments MDA5 signalling and the risk of clinically amyopathic dermatomyositis. Ann Rheum Dis 77:602-611,2018
3. Sone J, Mitsuhashi S et al Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 51:1215-1221, 20191)
4. Honda S, Kochi Y et al. Association of Polygenic Risk Scores With Radiographic Progression in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2022, AOP.