
Research at TMDU - Prominent Researcher
PDF Download
- Structural physiology of membrane proteins by cryo-electron microscopy

Distinguished Professor,
Cellular and Structural Physiology Laboratory (CeSPL),
TMDU Advanced Research Institute
A: The starting point of this research came out of a simple desire to understand human capacities at the molecular level. Membrane proteins are important for the function of nerve cells, but lipid membranes are only about 5nm (50Å) thick. To determine the molecular structure of such very thin samples, electron beams are more suitable than X-rays, because they interact much more with the samples, which is why I studied electron microscopy.
The pioneers of electron crystallography were Richard Henderson and Nigel Unwin, and they first determined the structure of a membrane protein, bacteriorhodopsin (bR), at 7Å resolution in 1975. When I began researching electron microscopy in the late 1970s, biological samples would get severely damaged by the electron beams, and high-resolution analysis was difficult. After various attempts, we found that if the temperature was reduced to 8K or less, sample damage caused by electron beams could be reduced to about 20 times less than at room temperature. Since then, we have made improvements for the eighth generation, which is currently under development. In 1997, using this type of electron microscope, we successfully determined the structure of bR at 3Å resolution.
A: One example is the aquaporin (AQP) family, which are known to work as water channels. The AQP family exists in all organisms, from bacteria to humans. A human has 13 different AQPs that function in various parts of the body.
AQP1 passes 3 billion water molecules per second, but does not pass ions or protons (H+). If ions pass, the ion channel cannot function, and if protons pass, the pH in the cell will change and cause dysfunction of the cell. However, since water molecules are connected by hydrogen bonds, protons could easily pass through the channel through the connection. The mechanism of such rapid water permeation and high water-selectivity of AQP was a big mystery.
We elucidated the molecular mechanism of AQP1 by analyzing its structure and published it in 2000 (Fig. 1). In 2009, we succeeded in individually observing eight water molecules in the channel of AQP4 (Fig. 2), and this mechanism is now more reliable. This achievement was made possible by electron crystallography, which allows analysis of membrane proteins embedded in lipid membranes. When X-ray crystallography is applied to the membrane protein, the lipid membrane should be removed. But this operation changes the charge distribution within the sample, so the position of the water molecules cannot be determined accurately with X-ray crystallography, despite its higher resolution than electron crystallography.
By the way, AQP2, which plays an important role in the kidney among AQPs, was discovered by Dr. Shigeru Sasaki, who was at that time a professor of Homeostasis Medicine and Nephrology at TMDU (currently emeritus professor). We determined the following mechanism: When a human is thirsty, a hormone called vasopressin is secreted from the pituitary gland, and when it reaches the kidney, AQP2 in the cell comes out on the cell surface and absorbs water, which otherwise would be excreted as urine. APQ2 inhibitors have potential for use in the treatment of heart failure and edema in cirrhosis, and we have started collaborative research with Dr. Sasaki.
Fig. 1: Proposed hydrogen-bond isolation mechanism based on the structure of APQ1 at 3.8Å resolution.
The middle of the channel is narrow enough to allow only one water molecule to pass. The two aspartic acids (⊕ in the figure) form hydrogen bonds with the oxygen (red spheres) of the water molecules that have arrived, breaking the hydrogen bonds between the upper and lower water molecules.
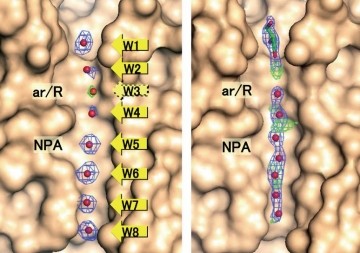
Fig. 2: APQ4 structure determined at 2.8Å resolution by electron crystallography (left), and structure solved at 1.8Å resolution by X-ray crystallography. Electron crystallography has lower resolution, but the eight water molecules (W1 to W8) in the channel are properly separated.
A: Yifan Cheng, who once was a post-doc in my lab, and his group analyzed the structure of the TRP receptor at high resolution using SPA with a cryo-electron microscope and a high-performance camera that can detect electron beams directly with CMOS. That was in December 2013. Their work triggered an explosion in SPA.
In 2017, three scientists received the Nobel Prize for their work on the development of cryo-electron microscopy and SPA. One of them, Arthur Henderson, explicitly mentioned at a press conference for the Nobel Prize, "Yoshi Fujiyoshi's contribution in Japan” (Fig. 3). The JEM-Z300CF cryo-electron microscope developed by JEOL using my patent is starting to be widely used for SPA.

Fig. 3: Prof. Fujiyoshi (left) and Dr. Richard Henderson (MRC, LMB) at the Nobel Prize award ceremony in 2017.
A: This term represents a discipline to clarify the mechanism of when we look, think and act, based on the structure of the molecule. For example, when we see an apple and judge that it is food, then reach out and carry it to our mouth — before each action, various molecules come into play, such as rhodopsin in the retina, ion channels in nerve cells, acetylcholine receptors at neuromuscular junctions, and so on. I have been attempting to determine the structure of all these molecules and understand human capabilities at the molecular level.
We use three types of cryo-electron microscopy to determine the structures — electron crystallography, SPA and electron tomography. This is the order of resolution, but the order of importance in a biological sense is the reverse. I've been doing analysis using electron crystallography for many years, but it took 18 years to analyze one of the receptors. Therefore, recently we have also been using SPA, which has made remarkable progress.
Currently, we are studying the mechanisms of acquired synaptogenesis, ion channel gating and tight junctions formed by claudins, in addition to water channels. In addition, SPA is helping us understand the unique gating mechanism of the gap junction, which is the center of the electrical synapse.
A: Since around 2000, "evidence-based drug development," which identifies target proteins and screens for compounds based on them, has become the mainstream method of drug discovery. However, even if promising compounds are found thorough screening, preclinical and clinical trials often reveal side effects, and the probability of success is only 1 in 30,000.
For this reason, “drug repositioning” to find other medicinal effects of already approved drugs is being actively conducted, but there is a risk that drug targets will be depleted. Also, in Japan, the same compound would have the same price, so pharmaceutical companies do not make much profit from repositioning their drugs.
Therefore, I came up with a strategy called “drug rescuing” to “rescue” compounds that are discarded during the drug development process. The promising way to implement this strategy is by determining the structure of the complex of target protein and the compounds. The structure will tell us the efficacy-related parts of the compound, allowing us to change the efficacy. It will also be useful to efficiently reduce side effects by modifying parts that are not related to drug efficacy.
Drug rescuing has come true because SPA has accelerated structural analysis, but cryo-electron microscopes, which are essential for SPA, are expensive and difficult to maintain. For this reason, we have launched a business venture offering structural analysis services, and have started operation in cooperation with TMDU. Specifically, we will set up our equipment at Tokyo University of Agriculture and Technology as a hub center and plan to build a system to remotely collaborate with pharmaceutical companies and universities via fiber-optic connections. We will also conduct joint research with pharmaceutical companies and others and hold workshops to share know-how for analysis.
A: At TMDU, in addition to the contributions I mentioned, I hope to use the network of overseas researchers I have built up to advance research at TMDU overall.
There is another reason I established the venture. The Medical Research Council, Laboratory of Molecular Biology (MRC, LMB) in the U.K. has only about 100 Principal Investigators but has so far produced 19 Nobel Prize laureates. I think that is possible here, because there is plenty of research funding, constructing a good environment where researchers can do their work without worrying about money. Moreover, a virtuous circle is forming. For example, Sir Gregory Winter, who developed the phage display method and won the Nobel Prize, donated a large amount of his patent revenue. I also have a tremendous dream of creating a system that allows researchers to conduct research by using the income we will earn through ventures.