
VCP, a causative gene of FTLD controls the common pathology of polyglutamine diseases (2013)
More than 10 years ago, we originally demonstrated that VCP binds to a polyglutamine-tract by a screening with yeast two-hybrid method (Imafuku et al., BBRC 1998). Thereafter, involvement of VCP in the pathologies and the binding between VCP in the pathologies and the binding between VCP and a polyglutamine disease protein (Ataxin-3) were reported by another group (Hirabayashi et al., Cell Death and Differ. 2001). These results suggested that VCP might be a molecule involved in multiple polyglutamine diseases. However, this question had not been directly addressed and the molecular mechanisms mediated by VCP in the pathogenesis had remained unclear. Beyond polyglutamine diseases, VCP, a multi-functional protein belonging to AAA ATPase family and carrying functions in membrane trafficking, ER protein degradation and DNA repair etc, is known to be a causative gene for a form of frontotemporal lobar degeneration (FTLD), IBMPD.
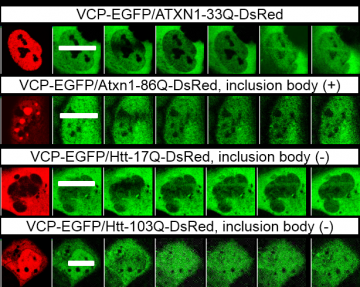
We hypothesized that there would be a common pathology among polyglutamine diseases mediated by VCP. In order to clarify impaired function of VCP in the common pathogenesis, we first tested the bindings between VCP and polyglutamine disease proteins (Ataxin1, Ataxin7 Hunthintin and AR). We found that VCP binds to all 4 types of polyglutamine disease proteins, and surprisingly both normal and mutant forms interact with VCP almost equally. The binding was dependent on polyglutamine tract sequence because the mutant forms of polyQ proteins lacking polyglutamine tract sequence did not bind to VCP. We also found the co-localizations of VCP and polyglutamine disease proteins in nuclear aggregates by immunostaining. Moreover, we found VCP was localized in the nucleus of neurons in contrast to cytoplasmic localization in the other types of cells. These findings suggested that VCP dysfunction in the nucleus of neurons might contribute to the common pathology. We reported that DNA double strand break (DSB) is involved in polyglutamine disease pathogenesis (Enokido et al., JCB 2010). Therefore, we tested the effect of polyQ disease proteins via VCP on DNA damage repair.
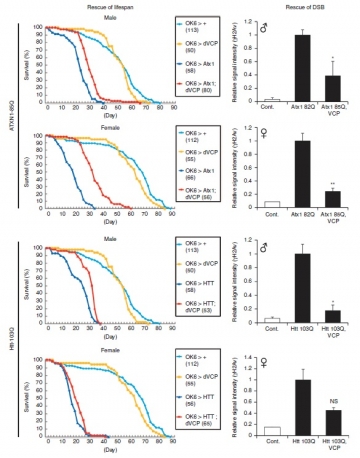
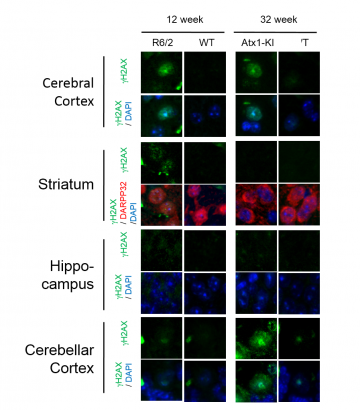
In drosophila and mouse models of HD and SCA1, signals of gamma H2AX or H2Av, DNA-DSB markers, were increased in neurons. Furthermore, VCP overexpression in transgenic models recovered the lifespan shortening and reduced gamma H2AX and H2Av signals. Micro irradiation method demonstrated mutant Ataxin1 and Huntingtin perturbed intracellular dynamics of VCP, and inhibited VCP accumulation to the DNA damage foci.
Collectively, our results suggested that any mutant polyQ disease protein can bind to VCP and will modify its dynamics. Consequently, DNA repair function is impaired and DNA-DSB is increased (Fujita et al. Nat Commun 2013).