東京都難病診療連携拠点病院、IRUD 拠点病院として
広く稀少難病疾患患者さんを診療し、先端的な診断と治療を提供します。
稀少疾患は、患者数が1万人に5人未満の疾患、と定義されています。
しかし、その種類は最低6,000~7,000 以上と数が多く、欧州では全人口の12 人に1 人が稀少疾患に罹患しているとされています。稀な疾患で、多彩な症状を呈することも多く、診断が遅れたり、診断が難しかったりすることもあります。適切な治療の選択にも高度な知識が必要です。当病院では、稀少疾患の先端医療にあたる専門医師により、領域を超えた連携診療を行っております。
センター長のご紹介

| 科長 |
蘇原 映誠 -Eisei Sohara- |
| 専門医 |
日本内科学会認定 総合内科専門医
日本腎臓学会認定 腎臓専門医・指導医
日本透析医学会認定 透析専門医・指導医 |
| 専門分野 |
遺伝性腎疾患
腎臓内科疾患
透析医療 |
| 研究領域 |
遺伝性腎疾患・腎臓難病の診断システムと病態解明
遺伝性腎疾患・腎臓難病に対する先端的治療法の開発
慢性腎臓病
腎生理学(水電解質代謝・高血圧など) |
| 電話番号 |
【予約に関して】03-5803-4655 (地域連携室 初診予約担当) |
医療機関からの難病診療部の初診事前予約方法
(電話またはFAX にてスムーズに受診予約できます。)
1.電話・FAX
難病診療部の受診方法
次の番号に電話または申込書のFAXをお願いします。(
申込書はホームページからダウンロードできます。)
| 受付時間 |
8:30 ~17:00(土日祝日、年末年始12/29 〜1/ 3は除く) |
| TEL |
03-5803-4655(地域連携室 初診予約担当) |
| FAX |
03-5803-0285(FAX受信は24時間可能)
※時間外、休日等のFAX 受信分は翌診療日にご連絡させていただきます。 |
2.予約日の決定
「外来診療予約票」を原則20分以内にFAX にて返送いたします。
3.紹介状(診療情報提供書)
予約日の前診療日正午までに紹介状をFAXにてご送信ください。
4.予約日に受診
当日の持ち物
●紹介状(原本) ●保険証またはマイナンバーカード ●外来診療予約票
予約時間の45分前までに1階初診受付窓口(⑤番)にお越しください。
取り扱うおもな疾患に関する詳細説明
原発性免疫不全症(PID)
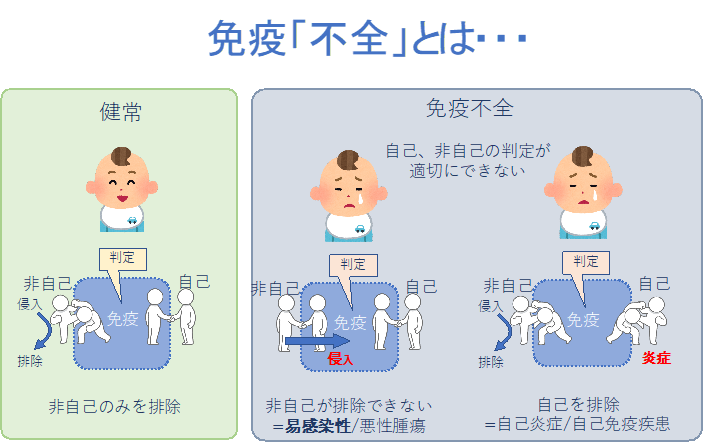
原発性免疫不全症候群(PID)は、免疫系のいずれかの部分に欠陥がある疾患の総称です。頻度は1万人に1人以上であり、従来考えられていた数よりも多いことが、近年わかってきました。決して稀な疾患ではありません。障害される免疫担当細胞などの種類や部位により400以上の疾患が含まれ、そのほとんどで責任遺伝子が同定されています。共通した症状は易感染性です。これは、免疫力の低下により、通常では感染することなく、通常の人に症状が出ない弱い病原体に対しても感染が起きることで臓器機能障害・症状が生じる状態や、軽症かつ一過性ですむはずの感染症が重症化したり慢性化したりするような状態を指します。疾患によりそれぞれ違う種類の微生物に対する脆弱性を示すため、病像は多彩です。また免疫機能の低下による自己免疫疾患、炎症性疾患、悪性腫瘍、アレルギー疾患の合併が多く、それらが前面にでることもあります。
PIDの症状は多彩であり、時にその診断は困難であり、未診断例も多く存在すると考えられています。PIDは軽症なものから重症なものまであり、治療も個々のタイプや重症度などによって異なります。ただ重症なタイプでは生後早期に骨髄や臍帯血による造血細胞移植を必要とします。こうした造血細胞移植は、PIDに対する根治的な治療法として行われますが、通常の悪性疾患などに対するものと異なる前処置やドナーの選定を行うことも多く、疾患の種類や患者の状態に応じたきめ細かい対応が必要となります。
当教室はPIDに対する専門医療を国内でも最も早くから行っており、PIDに対する造血細胞移植症例数は全国で最多です。また比較的新しい疾患概念であることから、成人科での専門医療が受けにくい疾患でもあり、成人の患者さんの治療も必要に応じて行っています。
PIDの適切な治療には、遺伝子診断を含めた正確な診断をつけることが前提となります。極めて多岐にわたるPIDを正確に診断するためには、高い診療技術と専門的検査が必要です。診断のために必要となる特殊な検査、例えば細胞生物学的解析、生化学的解析を行うための設備や、遺伝学的な検査のための次世代シークエンサーなども自施設に完備しており、必要に応じて他施設、例えばかずさDNA研究所などと協力しながら、迅速な診断ができるシステムを備えております。また遺伝子診療科と連携し、疾患に応じた遺伝相談も積極的に行っています。
さらに、当施設は日本免疫不全・自己炎症学会の中心的施設として、多くの専門施設からも診断や治療に関する相談を受けております。
性分化疾患(DSD)・先天性副腎過形成(CAH)
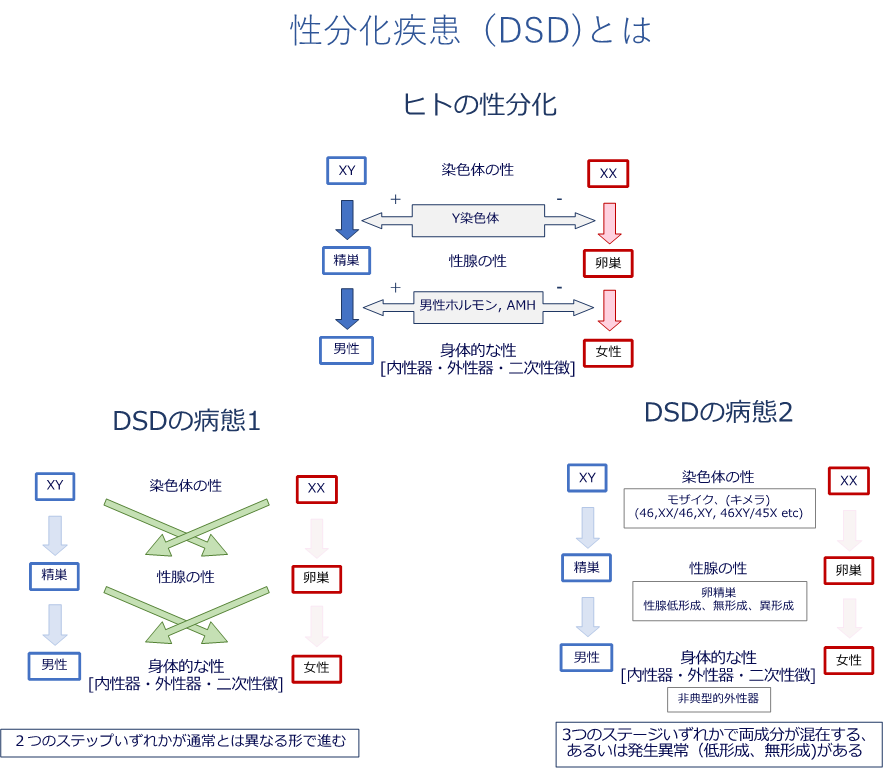
受精卵は男女の違いは明らかでありませんが、胎内で次第に男性女性の区別がはっきりし、生まれてくるまでの間に性別がはっきりとします。この過程を「性分化」といいます。性分化疾患(DSD)は、胎内での性分化が典型的に進まない状態を指します。生まれた時に外性器の特徴で性別を判別することが難しい、あるいは本来の性染色体の組み合わせ(核型)とは異なる性をもって生まれてくるお子さんなどが含まれます。
DSDにより、非典型的な外性器をもつお子さんが生まれた場合、他にはない難しいケアを求められます。それは、お子さんをどちらの性別で育てていくかという「社会的性の決定」です。これはDSD特有のものであり、DSDを扱う医療者が高い専門性を求められる要因のひとつになっています。
また、思春期の発来が通常通りこないことをきっかけに発見されることもあります。そうした場合、それぞれの性別に応じた治療と細やかなケアが必要となります。そうした際に遺伝学的検査も含めて、迅速に原因を診断する体制と経験が必要です。
当院は日本小児内分泌学会が指定するDSDの中核施設として多くの患者さんの診療を行っております。
先天性副腎過形成(CAH)
先天性副腎過形成は、副腎でコルチゾルというホルモンを産生する過程で必要な酵素が先天的に欠損するために生じる疾患です。この際、同時に性ホルモンの産生が異常になることがあり、生下時に非典型的外性器で男女の区別がはっきりしないことがあります。そのためCAHはDSDの一つでもあります。頻度は約1/20000程度とされています。
コルチゾルは我々の生命の維持に必須であるため、CAHでは生後早期に急激に具合が悪くなることがあります(副腎不全)。こうした状態を予め防ぐために、新生児マススクリーニングの対象疾患となっています。成人期以降の生殖能の維持も含めCAHの診療は専門性の高い医療が求められます。CAHの新生児マススクリーニングは今から30年ほど前に導入されましたが、東京都スクリーニングの導入は本学の指導のもと行われました。以降、当施設は国内でも最も患者数が多い施設の一つとして多くの患者さんを幅広く診療しています。
肺動脈性肺高血圧症
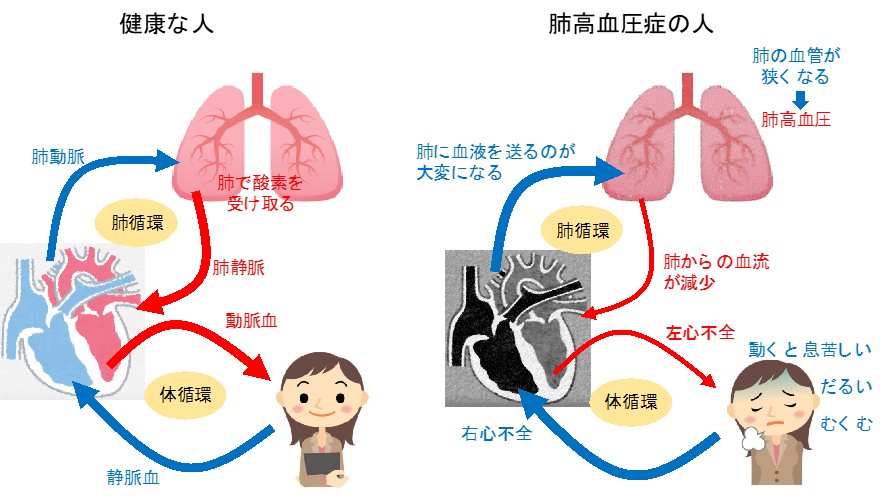
ヒトが生きるためには、大気中の酸素を「呼吸」を介して、心臓から肺に戻ってくる血液に取り込む必要があります。心臓から肺に血液を送るための血管を「肺動脈」といいます。この肺動脈の血液の流れが悪くなると、その血管の中の圧力(血圧)が上昇します。これを「肺動脈性肺高血圧症(PAH)」といいます。息切れ・疲労感・めまい・胸痛・血たんが主な症状で、飲み薬や点滴(肺動脈の血圧を下げる薬)で治療しますが、根本的な治療法はなく、最終的には心不全を起こし、長く元気に暮らすことができません。原因の一部は遺伝子の異常によることがわかっていますが、多くはその原因が解明されていません。PAHは成人に多いとされますが、小児でも認めることがあります。小児の場合、患者さんの将来性なども鑑みた上で、成人とは異なる治療の考え方が必要となります。当施設では、数少ない小児のPAHに対する国内でも有数の専門的な診療を行っています。重症肺高血圧症の患者さんに対しては、初期から複数の薬(肺高血圧標的治療薬)を併用(upfront combination therapy)し、治療効果が不十分な症例にはエポプロステノール持続静注療法を早期に導入しています。近年開発が進んでいる肺高血圧症の新しい標的治療薬も積極的に併用することで良好な治療成績をあげています。
なおさらなる詳細な情報については、以下のサイトもご参照ください。
- 東京科学大学病院 小児科 HP
-
https://www.tmd.ac.jp/med/ped/medical/specialisation_junkan.html
頭蓋骨縫合癒合症(craniosynostosis)
ヒトの頭蓋骨は縫合と呼ばれる線維性の結合組織によって連結されています。出生後から幼児期にかけて脳は顕著な成長を遂げますが、これに伴って頭蓋骨は外側に三次元的に拡大され、骨の断端には新しい骨が添加されます。さらに、頭蓋骨表面では骨リモデリングとよばれる骨の改造現象が起こります。これら複雑な機構が適切な時期に働くことは、頭頚部の健やかな成長発育にためには不可欠です。
脳を取り囲む頭蓋冠の縫合は、一般的に30歳ごろから徐々に骨癒合が起こって閉鎖します。一方、縫合が胎児期あるいは出生後早期に骨癒合する疾患を頭蓋骨縫合癒合症(craniosynostosis)と総称します。縫合の早期癒合によって頭部、顎および顔面の成長は三次元的に著しく阻害され、形態や機能に異常が引き起こされます。個人差や疾患差があるものの、癒合する縫合の部位や範囲によって、舟状頭、斜頭、短頭や三角頭蓋といった特徴的な頭蓋の形態異常が引き起こされます。また、顎や顔面の形態異常として、眼球の突出、両眼開離、顔面の非対称や反対咬合などが認められることも多く、重篤な不正咬合を有する患者さんも多いです。
頭蓋骨縫合癒合症は、様々な症候群に随伴する症候群性と、頭蓋縫合癒合症以外の症候を持たない非症候群性のものがあります。症候群性の頭蓋骨縫合癒合症には、後述するApert症候群やCrouzon症候群のほかに、Muenke症候群、Pfeiffer症候群やSaethre-Chotzen症候群等が知られています。当施設では、医科および歯科の様々な専門科の医療従事者が緊密な連携をとり、頭蓋骨癒合症の患者さんの治療にあたっております。
アペール症候群(Apert症候群)
頭蓋骨縫合早期癒合、特に冠状縫合が対称性に早期癒合することが多いです。また四肢の合指症は、本疾患の大きな特徴です。本疾患は、常染色体優性遺伝性の疾患で、出生率は100万出生に対して15~16人程度と、非常にまれな疾患であり、その多くが線維芽細胞増殖因子受容体(fibroblast growth factor receptor; FGFR)遺伝子における2種類の新生突然変異のどちらかが生じることで発症します。FGFRはヒトの生命活動に非常に重要な遺伝子の一つであり、全身の様々な組織で発現・機能しています。FGFRにアペール症候群型の変異が起こると、FGFRの正常な機能が損なわれ、様々な症状が起こるとされています。上顎骨の劣成長による反対咬合、開咬、重度叢生、永久歯の萌出障害、V字型の狭窄した歯列弓を示すことが多いです。遺伝子の変異部位によって、口蓋裂の発症率や合指症の重症度が異なることが知られています。また、口蓋に特徴的な上皮の肥厚が認められることがあります。クルーゾン症候群と比較して、クルーゾン症候群では顎顔面の骨格的な形態異常や不正咬合の重篤度が高いと報告されております。近年では、上気道の狭窄による睡眠時無呼吸症候群を併発することが知られ、呼吸状態の把握やケアの重要性が指摘されております。また、程度は様々ですが、前頭部~頭頂部にかけて骨欠損と、島状骨の形成が認められることも多く、指状圧痕とよばれる特徴的なレントゲン像が観察されることもしばしばです。
クルーゾン症候群(Crouzon症候群)
頭蓋縫合部早期癒合は様々な縫合でおこり、その部位や範囲も個人差があります。上顎骨の劣成長、浅い眼窩や眼球突出が認められます。本疾患は、常染色体優性遺伝性の疾患で、出生率は100万出生に対して5~16人程度と、非常にまれな疾患です。また、クルーゾン症候群患者数は、すべての頭蓋縫合早期癒合症患者数の4.5%を占め、患者さん全体の約7割が家族例で、3割が孤発例と報告されております。線維芽細胞増殖因子受容体(fibroblast growth factor receptor; FGFR)遺伝子での突然変異が生じることで発症しますが、変位部位は30か所以上が報告されています。上顎骨の劣成長による反対咬合、開咬、叢生、永久歯の萌出障害、V字型の狭窄した歯列弓を示すことが多いです。また、口蓋裂や高口蓋等が認められることがあります。クルーゾン症候群では不正咬合の程度に個人差があります。近年では、アペール症候群と同様に、上気道の狭窄による睡眠時無呼吸症候群を併発することが知られ、呼吸状態の把握やケアの重要性が指摘されております。
頭蓋骨癒合症患者さんに対する集学的治療
頭蓋骨縫合の早期癒合により、脳圧が亢進することによって様々な合併症が起こります。したがって脳圧を下げるために、前頭部を前方へ広げる頭蓋形成術が行われます。アペール症候群における四肢の合指症に対しては、合指の分離および指間の形成・皮膚欠損部への植皮術等が行われます。口腔内に口蓋裂がある場合は、口蓋形成術が行われます。これらの手術は患者さんの年齢や症状に応じて、適切な時期に行われます。また、不正咬合(反対咬合や開咬等)に対しては、矯正歯科、小児歯科、口腔外科や補綴科といった歯科の専門科による幼児期からの継続的な長期管理が必要とされます。特に顎骨の成長が盛んな思春期性成長期に不正咬合がさらに増悪するため、顎骨に対する手術(顎矯正手術)が必要と判断される場合もあり、その場合は成長終了後に施術されることが多いです。発音・嚥下・咀嚼といった口腔機能の改善、また顎顔面の審美性の改善を目的とした顎矯正手術には様々な術式があり、上顎骨や下顎骨の骨切りや上顎骨の骨延長などが行われます。また、言語聴覚士(Speech-Language-Hearing Therapist; ST)による訓練により、音声・言語機能の向上をはかります。
トリーチャー・コリンズ症候群(Treacher Collins症候群)
トリーチャー・コリンズ症候群とは頬骨と下顎骨の形成不全、外耳奇形、下眼瞼欠損、下睫毛欠損、毛髪位異常を特徴とする先天異常症候群です。発生頻度は2万5千から5万に1人であり、遺伝形式は常染色体優性をとりますが、一部(1%未満)は常染色体劣勢遺伝形式をとることもあります。原因遺伝子としては現在までにTCOF1、POLR1D、POLR1Cの3つの遺伝子が挙げられております。新生突然変異はおよそ6割を占めています。
トリーチャー・コリンズ症候群の典型的な症状として、眼瞼裂斜下、下眼瞼の欠損症いわゆるコロボーマなどの眼の症状、頬骨の形成不全に伴う頬部の平坦化、下顎骨の形成不全に伴う気道狭窄が主な症状です。また、40-50%に耳介の奇形および中耳腔の形成不全による伝音性難聴を有します。口腔内の所見として、口蓋裂を伴うこともあり、叢生および開咬、下顎後退などの不正咬合を呈します。
トリーチャー・コリンズ症候群患者に対する治療は、呼吸管理のため新生児期に気管切開、乳幼児期に下顎の骨延長術を行う場合があります。また口蓋裂を伴う場合、口蓋形成術が施行されます。難聴に対しては補聴器の使用または機能改善手術が施行されます。上下顎を含めた頬骨ならびに眼窩の再建も必要とされる場合もあり、出生直後から医科、歯科を含めた多領域の専門分野において集学的な治療が必要とされております。当施設においても、遺伝子診療科、産科、小児科、形成外科、耳鼻咽喉科、口腔外科、矯正歯科を中心とした専門診療科による集学的な治療が可能となっております。
鎖骨頭蓋異形成症(Cleidocranial dysplasia)
鎖骨頭蓋異形成症は、ヒトの骨の形成に関与する遺伝子の異常により、全身の骨や歯の形成に異常が生じる疾患です。頻度は100万人に1人と非常に稀ですが、常染色体優性遺伝様式をとるため、ご家族にこの疾患の方がいらっしゃる場合は発症の頻度は高くなる可能性があります。
主な骨の症状としては、鎖骨がないあるいは部分的に欠けている、大泉門がなかなか閉じないといったことがあり、身長もやや低めです。歯の症状としては、過剰な永久歯が顎の骨の中に存在していたり、大人になっても乳歯がなかなか抜けずに永久歯が顎の中にとどまっているといった問題が生じます。このような骨や歯の問題に対しては、出生直後から医科・歯科を含めた多領域の専門分野において集学的な治療が必要とされ、当施設では医科・歯科が連携しながら多くの本疾患患者の治療に当たっております。
家族性変性近視
1. 家族性変性近視とはどんな近視?
変性近視は、網膜への強い損傷が原因で、主に中年期以降から、視覚障害を生じる近視の深刻な形態です。網膜は、カメラで言えば「フィルム」のように機能する、目の奥の神経組織の層です。画像をキャプチャして脳に信号を送信します。変性近視による失明は、日本を含むアジア諸国における失明の一般的な原因です。変性近視の中に、常染色体優性遺伝に則り、明らかな家族集積性を示す予後不良な一群があり、「家族性変性近視」と呼ばれています。
2. どうして家族性変性近視になるの?
近視の人は、近くの物体をはっきりと見ることができますが、遠くの物体はぼやけて見えます。これは、近視の目が、通常よりも前後に長く伸びているために生じます(図1)。変性近視では、近視の程度が非常に強くなります。これは変性近視では、生涯にわたり目の強膜(目の白い部分)が前後に伸び続けるためです。変性近視では、はじめに、網膜の後ろにある脈絡膜と呼ばれる、網膜や強膜を栄養する重要な血管組織の層が薄くなり、やがて消失していきます(図2)。このため、加齢とともに、徐々に網膜に深刻な障害が生じるようになります。また、強膜は加齢とともに薄くなり、やがて正常な眼球の形を保つことができなくなり、『後部ぶどう腫』とよばれる、いびつな目の壁の突出を、目の後方に生じるようになります(図1)。家族歴のある人は、この状態を発症するリスクが高くなります。家族性変性近視では、何らかの遺伝子の異常により、眼球の壁を構成する組織に異常が生じる、もしくは眼球の伸びを制御するシグナルを出す神経網膜に異常が生じると推察されます。
3. 家族性変性近視はどのように管理する?
変性近視では、変性近視特有の様々な目の合併症が、中年期以降に生じますが、家族性変性近視では比較的若いうちから眼底に病変をきたすことがあります。これらの合併症を的確に診断することが、適切な治療を行うために必要です。以下に変性近視の代表的な合併症を示します。
- 3-a. 単純型黄斑部出血 (図3A)
目が伸びることで、網膜と脈絡膜を隔てるバリアのような働きをしているブルッフ膜という膜に、亀裂が入ることがあります。これによって、脈絡膜の毛細血管が障害されて出血が生じ、歪みや視力低下の原因となります。
- 3-b. 近視性脈絡膜新生血管 (図3B)
ブルッフ膜の亀裂を通って、脈絡膜から新生血管(しんせいけっかん)という病的な血管が網膜に入り込んで増殖してしまう病態です。変性近視で、中心視力が障害される最大の原因であり、血管内皮増殖因子(VEGF)を阻害する抗体を眼内に注入する治療を行う必要があります。
- 3-c. 近視性牽引黄斑症 (図3C)
目の壁が前後方向に伸びる際に、伸びきれなくなった網膜が分離してしまうことがあります。網膜の分離が限界に達し、分離では収まらなくなった場合に、網膜が剥がれはじめ、最終的に網膜剥離に至ります。
- 3-d. 近視性緑内障様視神経症 (図3D)
目が異常に伸び続けることで、視神経が変形して、網膜からの神経線維が、視神経で機械的に断絶するなどの障害を生じます。このため緑内障のように、視野が加齢とともに狭くなりますが、自覚症状がないことも多く、定期的な視野検査が必要です。
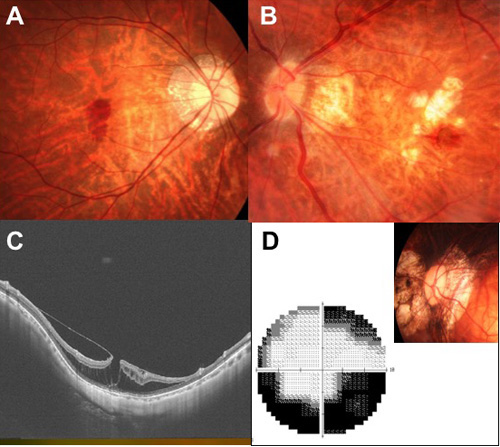
図1. 正常の目と変性近視の目の形の比較
目は前後方向に伸び、『後部ぶどう腫』と呼ばれる目の壁の突出(点線)をみとめる。
図2. 正常の目と変性近視の目の脈絡膜の厚さの比較
正常目では、脈絡膜に十分な厚みがある(両矢印)が、脈絡膜が非常に薄くなる。
図3. A. 単純型黄斑部出血、B. 近視性脈絡膜新生血管、C. 近視性牽引黄斑症、D. 近視性緑内障様視神経症
バージャー病
1. バージャー病とは
たばこを吸う20才代から40才代の男性(女性は少ない)にみられる代表的な手足の動脈の病気です。しかも徐々にと血管がつまってくるために、ほっておけば足が腐ってしまう病気です。別名として閉塞性血栓血管炎と言われることがあります。
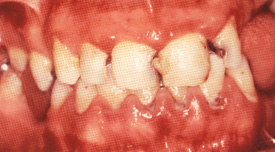
病気は、足趾(あしゆび)や手指にチアノーゼ(皮膚が濃い青紫色になること)や強い痛みがおこり、時間ともに皮膚が剥げたような潰瘍、腐った壊死と呼ばれる状態になります。ついには足だけの切断、ひどくなると膝関節の15cm位下での切断になることがあります。静脈にも炎症を起こすこと、たばこをやめると病気が進行しないことなど特徴があります。しかし依然原因不明の難病であり、国の特定疾患に指定されています。
アメリカ人のバージャー氏が1900年初頭研究論文を多数発表したことから彼の名があります。我が国では、1990年頃より発症患者さんが減少しています。欧米ではすでに我が国より早くから減少していますが、南および東アジアには依然多く、対策が急がれている血管の病気です。
われわれは、バージャー病にかかるということは、たばこによる歯周病の悪化に引き続く歯周病菌の血管感染によるものではないかと考え、幅広く研究を続けています。
2. 本学におけるバージャー病研究
この難病の原因の解明と予防法や治療法の開発のために、東京医科歯科大学ではバージャー病共同研究グループを組織して、歯周病学分野石川烈教授と当科前教授岩井武尚を中心に研究を進めてきました。
バージャー病患者の口腔内と患部の血管を調べて、歯周病とバージャー病との関連について検討しました。その結果、全てのバージャー病患者は歯周病と診断され、その程度はいずれも中等度から重症でした。また、患部の血管試料のほとんどから歯周病菌が検出されました。それに対して、正常血管の試料からは歯周病菌は全く検出されませんでした。
この発見は、今まで原因不明であったバージャー病と歯周病の関連を示した世界で初めての成果で、米国の血管外科専門誌 Journal of Vascular Surgery 41巻2005年7月号に 「Oral bacteria in the occluded arteries of patients with Buerger disease」 (バージャー病患者の閉塞動脈内に口腔細菌) と題して発表されます。
この発見によりバージャー病の原因や悪化が口腔内の細菌特に歯周病菌によるという可能性が強く示され、バージャー病の予防法や治療法の開発のための大きな手掛かりが得られました。
この成果について、2005年7月1日付けで各種報道機関において報道されました。
3. 今まで研究でバージャー病についてわかっていること
現在までにクラミジア肺炎菌、サイトメガロウイルス、ヘリコバクタピロリ菌、それに今回証明した歯周病菌がバージャー病以外の動脈病変からも見つかっており、そのことは当科がまとめた報告(ヨーロッパの学術雑誌)も含めて世界中から報告されております。
Dr Buerger自身また日本の研究者(Haga E)も含めて、急性期の病理所見などからなんらかの細菌感染(今回最も多かった歯周病菌と同じスピロヘータ属の梅毒、1928年Dr Allenの口腔内細菌説など)を強く示唆しておりました。バージャー病と喫煙は、強い因果関係があることはすでに証明されておりますし、喫煙により歯周病が悪化する事実に関しても多くの報告があります。社会的、経済的に安定し、さらに口腔内ケアーの行き届いた国ではバージャー病が減少しているという現実があります。バージャー病が減少している我が国においても口腔内ケアーの改善は、各年代において厚生労働省の資料でも明らかになっております。歯周病菌は血栓をつくりやすく、内皮細胞内に侵入するなどの事実がすでに報告されています。歯周病菌のPCR法による検出法は、キット化されており、確立された感度の高い検査法であります。歯周病菌は酸素を嫌う嫌気性菌であり、かつ口腔内に常在する共生菌とよばれる弱い菌であります。かつ、培養や抗菌薬治療に抵抗するきわめて扱いにくい細菌の一種であるとされています。
間質性肺炎(肺線維症)
肺は肺胞という小さな風船状の袋がたくさん集まってできており、私たちはこの肺胞の壁を通じて酸素を取り込んでいます。間質性肺炎とは、この肺胞の壁に炎症や損傷が起こる病気の総称です。このような炎症や損傷は徐々に肺胞の壁を厚く硬くすると考えられています。病状が進行すると肺胞の壁が厚くなり酸素を取り込みづらくなります。また肺が硬くなることにより深呼吸がしづらくなります。肺胞の壁が硬くなることを線維化と呼び、一度線維化した部分は元の軟らかい肺に戻ることはありません。
分類
間質性肺炎は原因によって以下のように分類されています。原因によって治療が異なることがあるため、問診、身体診察、血液検査、胸部CT、内視鏡検査や組織検査などの検査を行って原因の検討をします。肺の組織検査が正確な診断に必要なことが多いですが、組織検査には危険も伴うため、施行の是非は慎重に相談します。
-
自己免疫の異常によるもの:関節リウマチや強皮症といった膠原病は、自己の免疫が誤って自分の体の一部を攻撃してしまう疾患です。膠原病の多くは皮膚や関節に症状が現れますが、肺にも症状が出てしまうタイプがあります。当院では膠原病内科と連携してこのタイプの間質性肺炎の診療を行います。
-
じん肺・石綿肺:金属やアスベストなどの粉塵を吸入することで起こる間質性肺炎があります。この診断には職業や居住地などの情報が必要になります。
-
アレルギーによるもの(過敏性肺炎):鳥の糞や羽毛、家庭内のカビなどにアレルギーをお持ちの方もいらっしゃいます。これらの細かい物質を吸入すると肺の中でアレルギー反応が起き、肺胞の壁が徐々に厚くなると考えられています。当院はこのタイプの間質性肺炎の診断・研究では国内有数の施設となっています。
-
薬剤・放射線によるもの:服用した薬剤、サプリメントの副作用として間質性肺炎が起きてしまうことがあります。また癌の治療の際に受ける放射線照射により起こる間質性肺炎もあります。
-
遺伝子異常によるもの:非常に稀ですが、遺伝的な影響で間質性肺炎を来すこともあります。ご家族に間質性肺炎の方がいらっしゃる場合は、担当医にお申し出ください。
-
原因不明のもの(特発性):原因が分からないタイプを特発性間質性肺炎といいます。いまだ間質性肺炎の半数以上が原因不明にとどまっています。特発性間質性肺炎も組織病理学的パターン別に細分化されています。このなかでは特発性肺線維症(IPF)が80~90%と最も多いとされています
治療
- 特発性肺線維症(IPF)の治療
間質性肺炎の進行のスピードは患者さんによって大きく異なります。進行するIPFの患者さんには肺胞壁が固くなるのを和らげる薬(抗線維化薬と呼びます)の服用をおすすめしています。進行するIPFの患者さんは5年後の生存率が30%程度とされ、癌と同程度に低いという報告もあり、間質性肺炎の疑いがあると指摘を受けた場合には呼吸器内科の専門施設への受診を一度はお勧めします。抗線維化薬は病気の進行を緩やかにできる場合がありますが、効果には個人差があります。現在使用できる抗線維化薬にはピレスパ®、オフェブ®があります。
- その他の病型の間質性肺炎の治療
IPF以外の間質性肺炎では、多くの場合ステロイド薬(副腎皮質ホルモン薬)や免疫抑制剤が用いられます。IPF以外でも抗線維化薬が使用できる病型が徐々に増えておりますので、担当医とご相談ください。
- それ以外の治療
風邪などをきっかけに急激に病状が悪化することがあります。これを急性増悪(ぞうあく)と言います。急性増悪が起きると致死率が50%を超えるとも言われています。急性増悪を確実に防ぐ方法はありませんが、日常の手洗い、うがいを徹底するとともに、肺炎や季節性インフルエンザのワクチンを受けておくことが推奨されます。
特発性間質性肺炎
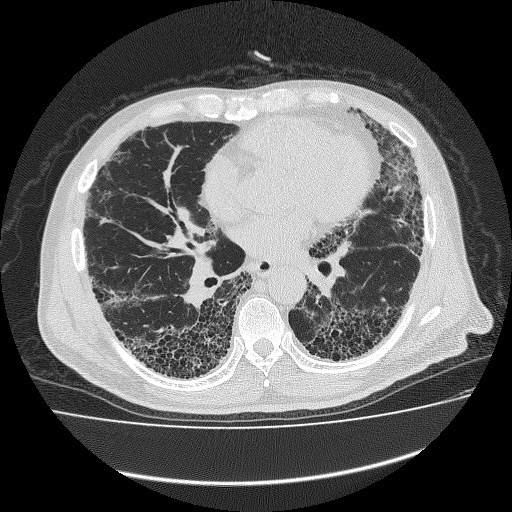
図1
特発性とは「いまのところ原因がわかっていない」ことを意味しています。この原因不明の間質性肺炎の総称を特発性間質性肺炎と呼んでいます。(間質性肺炎の項目もご参照ください。)特発性間質性肺炎はその肺病理組織のパターンにより細分化されています。このなかでは特発性肺線維症(IPF)が80~90%と最も多いとされています。胸部CTで肺が蜂の巣状に壊れる像を呈することが特徴です(図1)。
特発性肺線維症(IPF)の治療
間質性肺炎の進行のスピードは患者さんによって大きく異なります。進行するIPFの患者さんには肺胞壁が固くなるのを和らげる薬(抗線維化薬と呼びます)の服用をおすすめしています。進行するIPFの患者さんは5年後の生存率が30%程度とされ、癌と同程度に低いという報告もあり、間質性肺炎の疑いがあると指摘を受けた場合には呼吸器内科の専門施設への受診を一度はお勧めします。抗線維化薬は病気の進行を緩やかにできる場合がありますが、効果には個人差があります。現在使用できる抗線維化薬にはピレスパ®、オフェブ®があります。
その他の特発性間質性肺炎の治療
IPF以外の特発性間質性肺炎では、多くの場合ステロイド薬(副腎皮質ホルモン薬)や免疫抑制剤が用いられます。
慢性過敏性肺炎
慢性過敏性肺炎は間質性肺炎に含まれる疾患の一つです。(間質性肺炎の項目もご参照ください。)アレルギーによっておこる間質性肺炎で、鳥の糞や羽毛、家庭内のカビなどにアレルギーをお持ちの方が、これらの細かい物質を吸入すると肺の中でアレルギー反応が起き、肺胞の壁が徐々に厚くなると考えられています。当院はこのタイプの間質性肺炎の診断・研究では国内有数の施設となっています。治療にはステロイド薬(副腎皮質ホルモン薬)や免疫抑制剤などが用いられますが、最も重要なことはアレルギーの原因物質を遠ざけることです。
肺胞蛋白症
肺を膨らませるために必要な物質であるサーファクタントが肺内に過剰に蓄積されてしまう疾患です。サーファクタントを処理しているマクロファージが免疫の異常などで正常に働かなくなることから起こると考えられています。過剰に蓄積したサーファクタントにより、息切れが生じます。肺内に生理食塩水を注入し、過剰なサーファクタントを洗い流す治療が行われています。
リンパ脈管筋腫症 (LAM)
LAM細胞と呼ばれる異常な細胞が肺などで増殖する疾患です。ほとんどは妊娠可能な女性に発症します。LAM細胞が増殖すると肺内にのう胞といわれる穴が生じます。これが破れると気胸(肺に孔があいて空気が漏れる病気)を起こします。のう胞の形成が進行した場合は息切れが生じます。結節性硬化症に伴って発生する場合と単独で発生する場合があります。治療には肺機能の低下を防ぐ効果のあるシロリムスという薬剤が用いられます。
若年発症型両側性感音難聴
鼓膜および耳小骨を介して内耳に伝えられた音は、内耳に存在する有毛細胞という特殊な感覚細胞により電気信号に変換されます。電気信号に変換された音情報は聴神経を介して脳へと伝えられます。主に内耳および聴神経の障害が原因で起こる難聴を感音難聴と呼びます。原因不明で生じた両耳の感音難聴がゆっくりと進行する疾患を総称して特発性両側性感音難聴と呼びますが、特発性両側性感音難聴のうち、比較的若年(40歳未満)で発症し、難聴を引き起こす原因遺伝子変異を有することが明らかになったものを若年発症型両側性感音難聴と呼びます。
診断には、各種の聴覚検査や、その他の感音難聴を引き起こす疾患を除外するための画像検査に加え、遺伝学的検査が必要となります。当院では、これまで多くの特発性両側性感音難聴および若年発症型両側性感音難聴の患者さんの診療を行ってまいりました。
難聴の進行を抑える有効な治療法は確立されていませんが、当院では各患者さんの聴力に応じて、補聴器、残存聴力活用型人工内耳、従来型人工内耳などの装用により、きこえを改善できるようサポートしております。
メニエール病
難聴や耳鳴の出現とともに回転性のめまい発作を反復する疾患です。発症の原因は不明ですが、内耳に水ぶくれ(内リンパ水腫)が生じることが病態と考えられています。難聴は発症早期には低音域を中心とした感音難聴であり、めまい発作の軽快とともに改善することが多いですが、めまい発作を繰り返すとともに難聴は進行し不可逆性となります。両側性に発症することが多いことも本疾患の特徴です。
診断には、純音聴力検査や眼振検査に加え、内リンパ水腫の存在を推定する蝸電図検査、内リンパ水腫を可視化して確認する内耳造影MRI検査などを行います。
治療は発作予防をめざした生活指導や薬物治療が中心になります。日常生活では、適度な運動、規則正しい生活、十分な睡眠の確保などが重要です。代表的な薬物療法として、内リンパ水腫の軽減を期待できる利尿剤の内服が挙げられます。さらに、当院では、難治例を対象に、中耳加圧療法、内リンパ嚢開放術なども積極的に行っており、多くの治療経験があります。
遅発性内リンパ水腫
何らかの原因により片方の耳に高度の感音難聴を生じた患者さんにおいて、その高度難聴発症から数年から数十年経過した後にメニエール病様の繰り返すめまい発作、あるいは反対の耳に変動性の難聴を発症する疾患です。発症の原因は不明ですが、メニエール病と同様に内耳に水ぶくれ(内リンパ水腫)が生じて発症すると考えられています。
先行する一側性の高度感音難聴が存在する患者さんにメニエール病様のめまい発作を生じた際に本疾患を疑い、メニエール病の診断と同様に、各種の聴力検査、眼振検査、内耳造影MRI検査などの結果をもとに診断を行います。
治療はメニエール病と同様に発作予防をめざした生活指導や薬物治療が中心となります。難治例に対しては、中耳加圧療法のほか内リンパ嚢開放術やゲンタマイシンの鼓室内注入による選択的前庭機能破壊術を行いますが、一側の耳に高度難聴が存在することを考慮し、患側を慎重に判断した上での治療方針決定が重要になります。
無汗(低汗)性外胚葉形成不全症
無汗(低汗)性外胚葉形成不全症(anhidrotic ectodermal dysplsia)は毛髪、歯牙、爪、汗腺の形成不全を特徴とする遺伝性疾患です。主要な臨床徴候は、無汗(低汗)症など皮膚と付属器の形成不全による症状及び特徴的顔貌であり、その症状は永続的で進行はしません。
原因
低(無)汗性外胚葉形成不全症は、X連鎖劣性、常染色体優性または常染色体劣性の遺伝形式を示します。
- [X連鎖劣性遺伝性低汗性外胚葉形成不全症]
X連鎖劣性遺伝性の本症の責任遺伝子は、Xq12-q13.1に局在するectodysplasin A(EDA)です。現在までに、本症の原因として200種類以上のEDA遺伝子変異が報告されています。
- [常染色体優性・劣性遺伝性低汗性外胚葉形成不全症]
常染色体遺伝性の本症は、2q13に局在するEDA receptor (EDAR;別名DL)遺伝子または1q42.3に局在するEDAR-associated death domain(EDARADD)遺伝子の変異によって発症します。現在までに、本症の原因として5種類のEDARADD遺伝子変異が同定されています。EDA-A1、EDARおよびEDARADDは、外胚葉の形成に重要なシグナル伝達系(EDARシグナル)の主要構成分子であり、いずれの遺伝子に変異が生じても同様の臨床像を呈するとみられます。
症状
無汗(低汗)・疎毛・歯牙の低形成の3主徴を呈し、汗に関連する症状としては汗腺の欠如ないし低形成のため発汗の欠如または著しい低下をおこします。そのため体温調節障害が起こり、高熱下でのうつ熱症状、熱中症などが繰り返し起き、知能発達遅延をきたす場合や、乳幼児などは死亡に至る場合もあります。発汗の低下により、皮膚は乾燥が強くアトピー性皮膚炎様を呈します。皮膚の乾燥から眼周囲の色素沈着や雛壁が幼少期からみられるなどの特徴的な顔貌を呈します。また唾液腺など粘膜分泌腺の低形成もあるため、肺炎などの易感染性、萎縮性鼻炎、角膜びらんなどの症状がみられます。頭髪・腋毛・眉毛・睫毛・陰毛などの体毛は欠如または細く疎であり、歯牙は円錐状、杭状の切歯を伴う低形成や欠如などを認め、義歯の装着などが必要になることがしばしばであります。広く突出した額、鼻が低く鞍鼻、耳介低位、口唇は厚く外反し下顎が突出しています。病理組織学的には、表皮および真皮に大きな変化は汗腺や脂腺を認めない所見がみられます。患者は身体的機能の問題を持つと同時に、外観上・整容的な問題、社会的な活動の制限をもつため、心理的ケアを含めた診療体制や社会的な環境の整備の理解が求められます。
治療法
根治的な治療法はありません。長期にわたる対症療法と生活指導が必要です。毛髪は全身的に疎で薄く、色が薄い。毛の性状は粗くねじれており、脆弱であるため、洗髪の際は髪に負担を与えないように、シャンプー等の洗剤を用いる場合はよく泡立てて用い、地肌をこすらず、十分時間をかけてすすぐようにします。
爪は肥厚と変色を伴い突出しながらのびることがあります。爪は脆くなることもあります。時に爪は欠損します。爪の成長は遅いことが多いのですが、切らずに放置すると、外的な刺激を受けやすくなり外傷につながるため、日頃から爪切りやヤスリなどを用いた手入れを行うことが必要です。爪上皮は易感染性のため、手を洗う際は留意します。
歯牙の発達異常(欠歯、円錐歯など)があり、歯牙エナメルも不完全です。うがいや歯磨きなど口腔内の清潔を保つよう指導します。口腔内乾燥症状は口腔内細菌叢に悪影響を及ぼし、齲歯の原因ともなり得ます。口腔内保湿剤や人工唾液の使用も有用なことがあります。摂食障害を防ぐためには早期に義歯等の対応が必要です。義歯の定期的な交換も摂食、顎骨の発達、そして美容的な観点から極めて重要です。
眼は涙液の減少に伴うドライアイは角膜障害や眼瞼炎のリスクになることから日頃の人工涙液による保湿を心がけます。
萎縮性鼻炎に伴い、鼻閉・悪臭を伴う鼻汁は生活の質に影響を及ぼすほか、摂食および肺炎などの呼吸器系の問題に発展する危険性があります。日頃から鼻洗浄など鼻粘膜を清潔に保つことが推奨されます。
エクリン汗腺の分布は疎か完全に欠失します。そのため発汗機能が著しく損なわれ、体温が適切に制御できないためうつ熱になります。うつ熱は痙攣など神経学的な異常を来すことがあります。体温の上昇から多飲となり、多尿・夜尿につながることもある。そのため、夏場のうつ熱対策の工夫は重要です。何よりも周囲の病状に関する理解、そして暑い時期のクーリング対策、エアコン設置(学校・職場など)、暑熱環境(職場、屋外、入浴など)からの回避が必要です。熱中症を発症したときには入院して点滴による輸液補給が必要となります。汗の減少は皮膚の乾燥を生じ、皮膚炎の原因になり得ます。皮膚が乾燥した際は適宜保湿外用薬等を用いるなどスキンケアを心がけます。アトピー性皮膚炎様の湿疹病変に対してはステロイド外用が必要となることがあり長期の療養が必要です。
予後
汗に関連する症状としては汗腺の欠如ないし低形成のため発汗の欠如または著しい低下をおこします。乳幼児期には体温調節障害が起こり高熱下でのうつ熱症状、熱中症などが繰り返し起き、知能発達遅延をきたす場合や、乳幼児などは死亡に至る場合もあります。しかし、成人になるとうつ熱を避ける努力をすれば予後は一般的に悪くありません。
- 要件の判定に必要な事項
-
-
患者数
約50-100人(全国大学病院アンケート調査では最低25家系が明らかになっています)
-
発病の機構
不明(外胚葉の形成に重要なシグナル伝達系(EDARシグナル)の主要構成分子の遺伝子に変異を認めます)
-
効果的な治療方法
未確立、熱中症、アトピー性皮膚炎様の症状、肺炎などの感染症には対症療法、接触障害には義歯等の対応をします
-
長期の療養
うつ熱を予防する対策、熱中症の治療、義歯装着、スキンケア、肺炎、鼻炎、乾燥性角膜炎、口腔内乾燥などに対し長期療養は必要です
-
診断基準
あり
-
重症度分類
無汗性外胚葉形成不全症ガイドライン(日本皮膚科学会)の重症度分類を用いて対象は重症以上とする
- 情報提供元
-
難治性疾患政策研究事業 「無汗性外胚葉形成不全症の病態解析及び治療指針の確立」研究班
研究代表者:東京医科歯科大学皮膚科 皮膚科教授 横関博雄
学会名:「日本皮膚科学会」 代表者:山梨大学学長 島田眞路 (現在 ガイドラインの審査中)
「日本神経学会」 代表者:京都大学神経内科学教授 髙橋良輔
「日本発汗学会」 代表者:鳥取大学生理学教授 横関博雄
特発性後天性全身性無汗症
発汗を促す環境下(高温、多湿)においても、発汗がみられない疾患を無汗症といます。まれな疾患で発症率は明らかでありません。無汗のため、皮膚は乾燥し、時にはコリン性蕁麻疹を合併することもあります。また、高温の環境下において体温調節ができず熱中症を容易に発症し、発熱、脱力感、疲労感、めまい、動悸さらには意識障害など重篤な症状が出現することもあります。このため、夏には外出できなくなるなどの生活の制限があり、QOL(生活の質)が著しく損なわれます。
その中でも特に、特発性後天性全身性無汗症(AIGA:acquired idiopathic generalized anhidrosis)と呼ばれる疾患は、現在診療ガイドラインが改定(自律神経 [日本自律神経学会誌]、印刷中)され指定難病となっています。特発性分節型無汗症とidiopathic pure sudomtor fairlure(IPSF)などに分類されますが、その病態はいずれも明らかにされていません。IPSFは血中のIgEが高値である他、エクリン汗腺のアセチルコリン受容体に対する自己免疫疾患である可能性が推測されています。一部は、全身性ステロイド投与により軽快することが知られていますが、一般に治療が困難な疾患です。
疫学
特発性後天性全身性無汗症(AIGA)の正確な疫学的調査による報告はなく、その有病率・罹患率は不明です。これまでの症例報告は100例程度で、稀な疾患と推測されます。しかし、暑熱で過ごしたり、激しい運動をしない限り、症状が明らかにならないため、発病に気付かなかったり、他の疾患、例えば無汗症を伴うコリン性蕁麻疹や無汗症を伴うアトピー性皮膚炎、などと診断を受けている症例もあると考えらます。従って、正確にAIGAと診断される症例が、全体のごく一部である可能性もあります。
症例報告のほとんどは日本からであり、有病率に人種差、地域差がある可能性があると考えられています。また患者のうち8割以上が男性で、性差が際立っています。発症年齢は10歳代から30歳代の若年に多いものの、幼児から70歳代までのあらゆる年齢で発症する可能性があります。
なお、国内での研究班が行った調査結果では、本邦大学病院神経内科、皮膚科94施設における過去5年間のAIGA患者総数は145例(男性126例,女性19例)で、発症率は有意に男性において高率でした。発症年齢は1歳~69歳で、好発年齢は10歳代~30歳代。平均年齢は30.3歳(男性31.0歳,女性22.7歳)でした。
原因
発汗神経から汗腺への神経伝達を担う物質は、通常の交感神経と異なり、アセチルコリンです。発汗神経活動は、バースト状にアセチルコリンが放出されることで行われており、呼吸運動にある程度同期しています。汗腺からの汗滴分泌は発汗神経活動に同期して拍出されており、発汗波と呼ばれます。AIGAでは、①発汗神経障害( Sudomotor neuropathy) ②特発性純粋発汗不全(Idiopathic pure sudomotor failure : IPSF) ③特発性汗腺不全( Sweat gland failure) の3つの病態が考えられています。
マイクロニューログラフィにより記録される汗腺支配の皮膚交感神経活動は、①発汗神経障害では低下していますが、②IPSFと③特発性汗腺不全の初期では、正常または亢進しています。したがって、②③は発汗を誘発する神経シグナルは正常であるか亢進しているものの、汗腺が応答しない状態にあると言えます。③特発性汗腺不全の場合には、汗腺自体に異常があるために発汗しませんが、②IPSFの場合は、発汗運動神経末端から放出されるアセチルコリンに対して、汗腺のコリン受容体が反応しないことにあると考えられる。若年男性に多く、疼痛・異常知覚やコリン性蕁麻疹を合併しやすいが、精神性発汗は保たれるます。これはコリン受容体に作用できない過剰なアセチルコリンによるものと考えられています。さらに血清IgE高値を示す症例が多く、早期ならばステロイド・パルス療法が著効することからも、自己免疫的機序が推察されています。発汗神経障害の障害部位には、(1)視床下部、(2)延髄、脊髄(3)交感神経節前、節後遠心性線維の可能性が示唆されるが、いずれも環境温の変化に対し、皮膚交感神経活動の発射活動に変化がみられません。特に視床下部性の障害に対しては、発汗閾値の低下のこともあります。延髄、脊髄の障害においては、通常は無汗以外の神経症状を伴います。交感神経節前、節後遠心性障害の障害においては、髄節性の障害を呈し、同時に血管収縮神経障害を多く合併しています。末梢皮膚血流量をレーザードプラー皮膚血流量計により計測し、通常ではみられる皮膚交感神経活動のバースト発射に反応してみられる皮膚血流量低下が、発汗運動神経障害においては見られません。一方、IPSFや特発性汗腺不全では、皮膚血流量の低下が正常に認められる。IPSFと特発性汗腺不全の間には、病歴に差があり、特発性汗腺不全の病歴はかなり長くなっています。このことからも、自律神経性ニューロパシーや発汗神経障害、あるいはIPSFによる無汗症の二次的変化に伴い、組織学的変性を起こす場合と、原発性に免疫学的破壊が汗腺に起こることにより無汗を呈する場合があると考えられるますが、この両者は現在においては鑑別ができません。したがって、特発性汗腺不全には、heterogenousな多くの病態が含まれる可能性があります。これに対してIPSFはかなり確立したhomogenousな疾患と考えることができます。AIGAの中でもIPSFの占める割合は多く、IPSFは狭義のAIGAといってもよいと考えられます。
症状
無汗/減汗(発汗低下)は全身の広範囲にみられるますが、発汗が一部残存することも少なくありません。特に頭部、顔面、腋窩、手掌・足底は発汗が残存しやすい部位と考えられています。罹患している方では、体温調節に重要な発汗が障害されるため、運動や暑熱環境でうつ熱を起こし、全身のほてり感、体温上昇、脱力感、疲労感、顔面紅潮、悪心・嘔吐、頭痛、めまい、動悸などがみられ、熱中症に至ることもあります。運動や暑熱環境で誘発される皮膚のピリピリする痛み・発疹(コリン性蕁麻疹)がしばしばみられます。一部に自然寛解する例もあるが、多くは慢性の経過をとります。
合併症
コリン性蕁麻疹
治療法
- 副腎皮質ステロイド薬
根拠は十分ではありませんが、多数の症例報告の知見から推奨される治療です。しかし、発症から治療開始までの期間が長い例、汗腺組織の変性がみられる例では反応が不良とする報告があり、発症早期に行うことが勧められます。ただし保険適用外です。
- 免疫抑制薬の内服
ステロイド・パルス療法が無効な例で試みる価値があります。ただし保険適用外です。
- 抗ヒスタミン薬
症状に応じた抗ヒスタミン薬の適宜増量投与を検討することができます。
- その他の治療法(紫苓湯内服とステロイド外用)
難治な例が多いことを考慮すれば試みてもよいが、いずれも確立していない。保険適用外である。
研究斑
- 研究代表者
- 横関 博雄 東京医科歯科大学大学院医歯学総合研究科皮膚科学分野 教授
- 分担研究者
-
佐藤 貴浩 防衛医科大学校皮膚科 教授
室田 浩之 大阪大学大学院医学系研究科情報統合医学皮膚科学 准教授
渡邉 大輔 愛知医科大学皮膚科 教授
中里 良彦 埼玉医科大学神経内科 准教授
朝比奈 正人 医療法人同和会神経研究所神経内科 主任研究員
岩瀬 敏 愛知医科大学第2生理学 教授
- 研究協力者
-
藤本 智子 東京医科歯科大学医歯学総合研究科 非常勤講師
宗次 太吉 東京医科歯科大学大学院医歯学総合研究科皮膚科学分野 講師
結節性硬化症
結節性硬化症は、全身の様々な場所に症状が出ますので、症状に合わせた治療が必要となります。複数の科で連携することが重要で、東京医科歯科大学では結節性硬化症診療チームを結成し、治療にあたっています。
結節性硬化症は全身の様々な場所に腫瘍ができたり、てんかんや自閉症などの症状が出る病気です。原因遺伝子であるTSC1、TSC2の異常によっておこる、常染色体優性遺伝形式の病気です。遺伝子の異常によって、血管や細胞の増殖を調整するmTORが暴走し、体の様々な細胞が過剰に増殖します。
結節性硬化症は、それぞれの症状に出現しやすい時期があるのが特徴です(図1)。胎生期から乳児期に出現する心臓の横紋筋腫、出生時より認められる皮膚の白斑、乳幼児期から出現するてんかん、自閉症、精神発達遅滞、顔面の血管線維腫、乳幼児期に問題になることの多いSEGAなどの脳腫瘍、眼底の過誤腫、小児期から思春期に著明になる腎の血管筋脂肪腫(AML)、20歳以上の女性に問題となることが多い肺リンパ脈管筋腫症(LAM)などがあります。
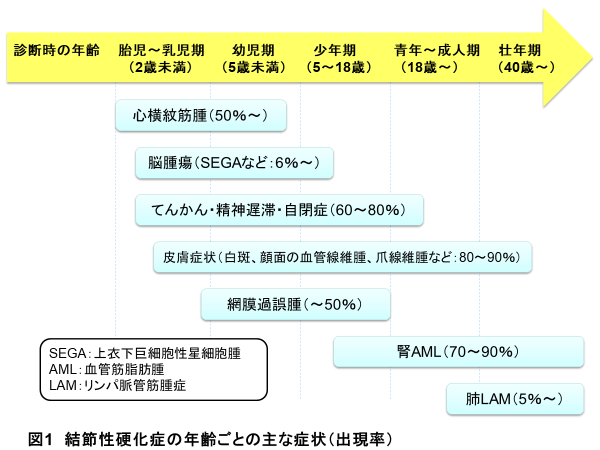
結節性硬化症診療チームは、下記の診療科によって結成されています。各診療科の紹介です。
小児科
(担当:森尾友宏、水野朋子)-
これまで根本的な治療はないとされてきましたが、mTOR阻害薬の登場で、治療法が大きく前進しました。小児科では主にてんかん、自閉症、脳腫瘍、心横紋筋腫の治療、管理を行っています。
てんかん:様々なタイプのてんかんを発症しますが、頭部を前屈させるタイプのてんかん(ウエスト症候群)を発症した場合は、治療に難渋することが多いです。抗てんかん薬、ビガバトリン、ACTH療法、ケトン食、mTOR阻害薬などで治療を行います。脳外科とてんかん外科手術を検討することもあります。
脳腫瘍:特に上衣下巨細胞性星細胞腫(SEGA)による水頭症が問題となることが多く、手術の他、mTOR阻害薬を使用する場合もあります。
心横紋筋腫:心不全、不整脈などの症状があれば、それらに対し適切な治療介入を行います。
小児科ホームページ»
脳神経内科
(担当:横田隆徳・西田陽一郎)-
結節性硬化症患者さんではてんかん発作を認めることがあり、発作頻度や重症度、発作型に応じた薬物治療を行います。薬物治療での発作コントロールが困難と判断した難治性てんかんの場合は、適切な時期に当院脳神経外科・てんかんセンターと連携して外科的治療のタイミングを計ります。
脳神経内科ホームページ»
脳神経外科
(担当:前原健寿・稲次基希)-
脳神経外科では、主に薬剤抵抗性てんかんの外科治療と上衣下巨細胞性星細胞腫(SEGA)の治療を行っています。結節性硬化症の患者さんでは、てんかんが薬剤抵抗性であることが多く、発作のコントロールに苦慮することがあります。私たちは、根治を目指す焦点切除術から、迷走神経刺激療法や脳梁離断術のような緩和術まで幅広く行っています。
脳神経外科ホームページ»
呼吸器内科
(担当:宮崎泰成・立石知也)-
結節性硬化症では、リンパ脈管筋腫症(LAM)と言われる疾患を主として肺に起こすことがあります。LAM細胞と呼ばれる異常な細胞が肺などで増殖すると、肺内にのう胞といわれる穴が生じます(これは肺のCTで見ることができます)。これが破れると気胸(肺に孔があいて空気が漏れる病気)を起こします。のう胞の形成が進行した場合は息切れが生じます。呼吸器内科では肺LAMに対する治療を担当します。
呼吸器内科ホームページ»
泌尿器科
(担当:藤井靖久・田中一)-
結節性硬化症の患者さんは、高率に腎腫瘍を有することが知られており、最も頻度の高いのは良性の血管筋脂肪腫ですが、腎細胞がんを発症することもあります。私たちは、私たちは、画像診断を駆使した血管筋脂肪腫と腎細胞がんの鑑別に取り組み、また手術適応となる場合には、最大限の腎機能温存を目指した無阻血腎部分切除術を施行しております。破裂の危険性のある大きな血管筋脂肪腫に対しては、選択的動脈塞栓術(放射線診断科と合同で施行)、あるいは薬物療法(mTOR阻害薬)を行っています。
泌尿器科ホームページ»
皮膚科
(担当:横関博雄・並木剛)-
皮膚科では、結節硬化症の皮膚症状である白斑・血管線維腫・シャグリンパッチ・爪囲線維腫に対しての診断および治療を行っています。特に顔面に生じる血管線維腫は審美的および美容的な側面からも悩んでいる患者さま多いですが、当科ではラパリムスゲル外用の経験が豊富であり外用による消褪傾向が見られることから、多くの患者さまから好評を得ております。またシャングリパッチや爪囲線維腫に対しては外科的切除や液体窒素療法などを試みており、近くにはCO2レーザーによる加療も予定しております。
皮膚科ホームページ»
眼科
(担当:大野京子・鴨井功樹)-
眼科では、眼病変(網膜過誤腫・網膜無色素斑)の診断や、治療薬であるビガバトリン使用時の眼における副作用の検出を、網膜電図などを用いて行います。
眼科ホームページ»
精神科
(担当:高橋英彦・高木俊輔)-
精神科では、結節性硬化症に良く認められる精神神経症状に対して包括的なアプローチを行なっています。結節性硬化症には自閉傾向、知的障害、攻撃性など多様な精神症状が出現し、結節性硬化症関連神経精神症状(TAND)とも呼ばれています。精神症状は患者さんそれぞれの違いが大きく、それぞれに異なる対応が必要です。また、結節性硬化症の患者さんは睡眠の質が悪く、そのため不眠と日中の眠気があることが多いと言われています。てんかんに対する抗てんかん薬治療を含めたこれらの精神神経症状に対して治療とケアを提供します。
精神科ホームページ»
遺伝性尿細管機能異常症
1.Bartter症候群/ Gitelman症候群
- <どのような病気か>
-
Bartter症候群/ Gitelman症候群は、腎臓でナトリウム(Na)を尿から体内に引き戻す(再吸収する)のに必要なNa輸送体タンパク(NKCC2やNCCなどの)遺伝子に生まれつき変異があり、尿中に必要以上のNaが漏れ出てしまうことで起きる病気です(図1)。一般的に体内のカリウム(K)もあわせて不足する状態となり低カリウム血症をきたし、血液がアルカリ性に傾く代謝性アルカローシスという状態になります。この病気の原因として、腎臓尿細管におけるNa調節に関わる7種類の遺伝子異常が知られています(下表)。近年ではこれらの疾患を総称して遺伝性(塩類喪失性)尿細管機能異常症と呼ばれます。
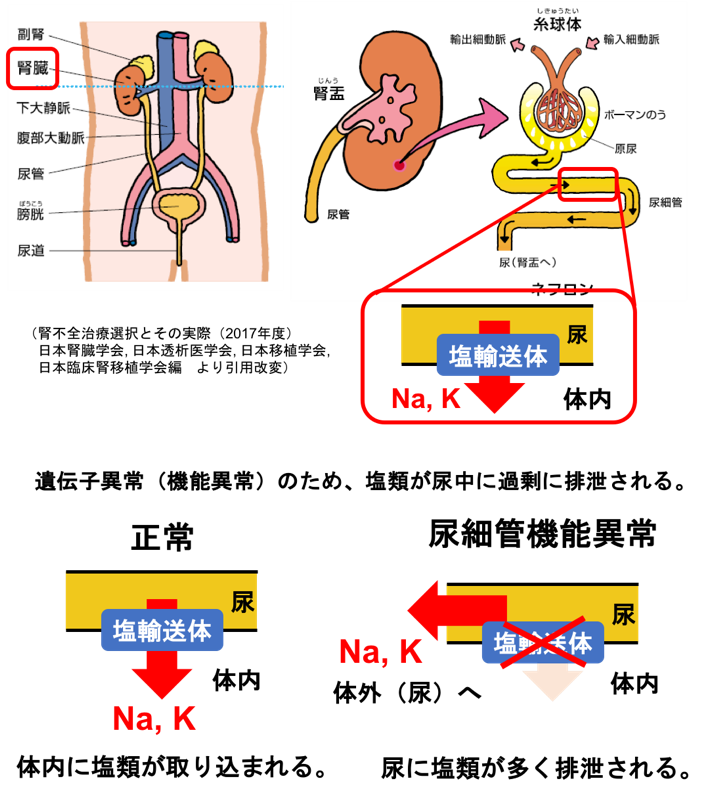
図1
症状や各種検査結果からこれらの原因遺伝子までを区別することは容易ではありません。当科では次世代シークエンサーという遺伝子解析機器を用いた網羅的遺伝子診断を行うことで、確定診断を行なっています。
- 症状
-
Bartter症候群は症状が強いことが多く主に新生児期から乳児期に診断され、低出生体重、多尿や成長障害、運動発達障害などの症状が出ます。Gitelman症候群はBartter症候群に比べて比較的軽症にとどまる場合が多く、症状は主に全身倦怠感、脱力や手足のしびれなどで、学童期以降・成人期に診断されることが多いです。Gitelman症候群は欧米の報告では4万人に1人程度で遺伝性尿細管機能異常症のなかでは最も多いとされていますが、日本人ではさらに高頻度で存在する可能性が指摘されています。
- 治療
-
低K血症に対する治療が主体となります。経口K補給薬の内服、K保持性利尿薬の内服、低マグネシウム(Mg)血症を合併する場合には経口製剤によるMgの補充などを行います。Bartter症候群は将来的に腎不全を多く合併しますが、Gitelman症候群では腎不全に至るケースは非常にまれと考えられています。
2.先天性腎性尿崩症
- どのような病気か
-
先天性腎性尿崩症は腎臓でできた尿を十分に濃縮することができず、薄い尿が多量に出る病気です。尿の量は抗利尿ホルモン(ADHやバゾプレシンとも呼ばれます)によってきめ細やかに調節されています。体内の水分調整は、ADHが腎臓の集合管という場所にある受容体(バソプレシン2型受容体;AVPR2)に作用して、水を通す分子である水チャネル(アクアポリン2;AQP2)を介して尿から水分が再吸収されることで行われています。腎性尿崩症は、AVPR2ないしAQP2の遺伝子変異により、ADHに対する腎臓の反応性が低下するために尿を濃縮することができない疾患です。本邦での正確な罹病率は不明ですが、海外の報告によれば10万人に1人程度と推定されます。乳児期早期に診断される例が多く、日本の報告では約7割が1歳未満で診断されており、6割は生後6ヶ月未満で診断されています。当教室では先の項目で記載した通り次世代シークエンサーによるパネル遺伝子診断を用いてAVPR2遺伝子とAQP2遺伝子を同時に評価しています。
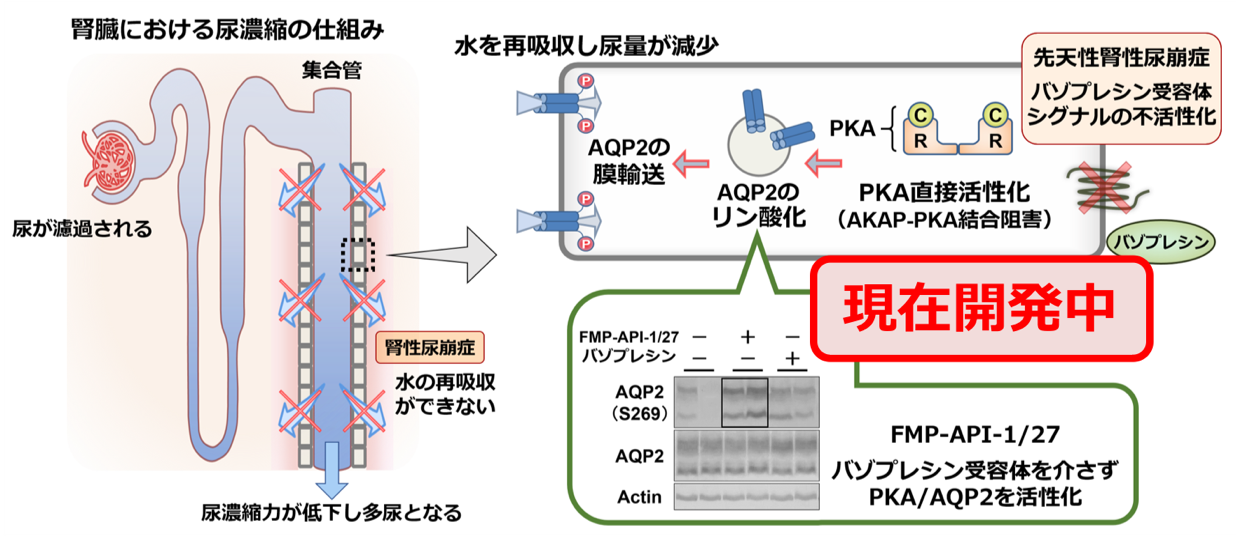
図2
- 症状
-
尿が濃縮できないことによる多尿(尿の過剰産生)と多飲(過度の口渇)です。年齢によって症状は異なります。
胎児期:母体の羊水過多
新生児期、乳児期:尿の量が多い(多尿)、水分をたくさんとる(多飲)、嘔吐、便秘、原因不明の発熱、体重増加不良、血液中の塩分であるナトリウムの濃度が高くなる状態(高ナトリウム血症)によるけいれん
幼児期~成人:尿の量が多い(多尿)、水分をたくさんとる(多飲)、大量に水分を摂取することによる食欲の低下、体重の減少
- 治療
-
根治的な治療を行うことは現時点では困難で、相対的に尿量を減少させることを目的として利尿薬(サイアザイド系利尿薬)や、ナトリウム摂取の制限、加えて腎臓の血流を落として尿量を減少させる目的でインドメタシンなどの非ステロイド系抗炎症薬(NSAIDs)が併用されることがあります。軽症の場合には抗利尿ホルモンの点鼻が有効な例があります。日常生活では水分摂取を制限することは避け、自由に飲水と排尿ができる環境をつくることが大切です。
また当教室では最近PKA, AKAPという分子に着目し尿濃縮の新しいメカニズムを明らかにしました。これらの分子をターゲットにした新たな治療は、これまで対症療法が主体となっていた腎性尿崩症に対して、全く異なった効果的な治療法となる可能性を秘めており、臨床応用を目指して研究を進めています。
3.その他の尿細管機能異常症
当科では上記疾患以外にも遺伝性低尿酸血症、Dent病、偽性低アルドステロン症(I型/II型)、Liddle症候群、尿細管性アシドーシス、Fanconi症候群などの尿細管機能異常症の網羅的遺伝子検査を進めております。
遺伝性腎炎
1.アルポート症候群・菲薄基底膜病
- どのような病気か
-
アルポート症候群(Alport症候群)は慢性腎炎(タンパク尿や血尿)、難聴、眼の合併症をおこす病気で、しばしば末期腎不全へと進行します。腎臓の糸球体という部分を構成するのに重要な蛋白である4型コラーゲンのα3鎖、α4鎖またはα5鎖をコードしている遺伝子COL4A3、COL4A4、COL4A5遺伝子のいずれかに異常がある場合に発症します。これらの蛋白は内耳や眼にも存在するため、難聴や眼症状を合併することがあるのはこのためです。国内での頻度は不明ですが、欧米からは5,000-10,000人に1人の割合で発症すると報告されています。遺伝性の疾患であり、ほとんどの場合、ご家族に同様の腎炎の患者さんがいらっしゃいます。しかし、約20%の患者さんでは家族歴がなく、遺伝子の突然変異により発症します。最も頻度が高いのはCOL4A5遺伝子の変異によるX染色体連鎖型アルポート症候群で、男性患者さんでは女性患者さんに比べて明らかに重症の症状を呈することが知られています。また比較的まれではありますが、COL4A3遺伝子やCOL4A4遺伝子の異常でも常染色体優性型や常染色体劣性型の遺伝をします。それぞれの特徴は以下の表の通りです。一方で菲薄基底膜病という診断名があり、これは血尿ないしは軽度のタンパク尿のみの症状が出ますが、腎不全は進行しないものを指します。原因となる遺伝子はCOL4A3/4A4で一部のアルポート症候群と同一ですが、基本的にはヘテロ接合性変異の型をとります。同じ変異でも症状の出方には個人差があり注意が必要です。当科では次世代シークエンサーという遺伝子解析機器を用いた網羅的遺伝子診断を行うことで、確定診断を行なっています。
| 遺伝形式 |
原因遺伝子 |
頻度 |
末期腎不全となる年齢 |
| X染色体連鎖型 |
COL4A5 |
80% |
男性 平均25歳
女性 40歳で12% |
| 常染色体劣性型常染色体劣性型 |
COL4A3またはCOL4A4 |
15% |
平均21歳(男女差なし) |
| 常染色体優性型 |
COL4A3またはCOL4A4 |
5% |
平均60歳前後(男女差なし) |
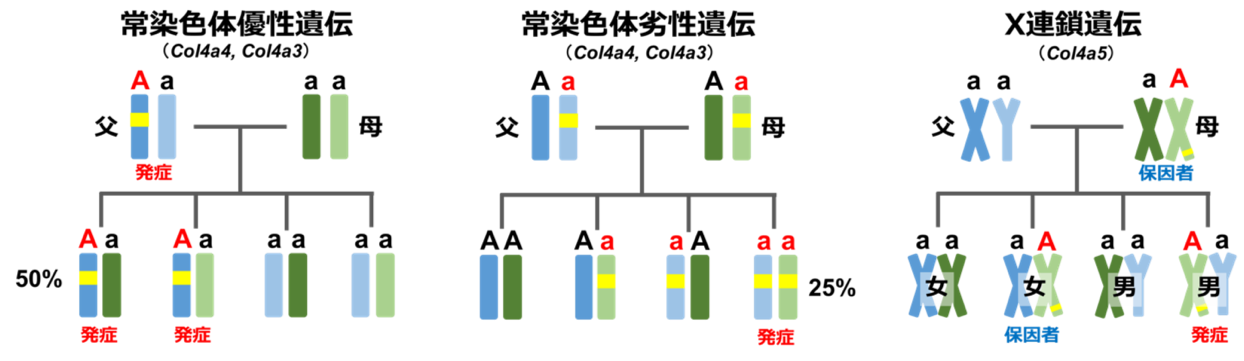
図3
- 症状
-
慢性腎炎:典型例では幼少期から血尿を認め、年齢とともに蛋白尿が出始める場合が多いです。最も頻度の高いCOL4A5遺伝子の異常に伴うX染色体連鎖型アルポート症候群では、男性では40歳までに約90%の患者さんが末期腎不全に進行します。一方、女性では40歳までに約10%の患者さんが末期腎不全へと進行します。末期腎不全へと進行した際は、透析や腎臓移植など、腎代替療法と呼ばれる治療が必要です。
難聴:生下時や幼少期に認めることはありません。しかし、最も頻度の高いCOL4A5遺伝子の異常に伴うX染色体連鎖型アルポート症候群では、男性ではほとんどの場合10歳以降に発症し、最終的には80%の患者さんで難聴を認めます。一方女性では20%の患者さんに認めます。
眼合併症:白内障や円錐水晶体などを認めることがあります。最も頻度の高いCOL4A5遺伝子の異常に伴うX染色体連鎖型アルポート症候群では、男性では約3分の1の患者に認めると報告されております。一方、女性においては非常にまれと考えられております。
- 治療
-
現在まで根治的治療法はなく、様々な対症療法を行いつついかに末期腎不全への進行を抑えるかに焦点が当てられております。尿蛋白を認める患者さんではアンジオテンシン変換酵素(ACE)阻害薬やアンジオテンシン受容体拮抗(ARB)薬による腎保護効果を期待した治療が実施されるのが一般的です。腎機能障害を認めない時期においては通常の日常生活を送ることができ、生活上の制限は必要ありません。
2.家族性腎臓病
(遺伝性巣状分節性糸球体硬化症[FSGS]、常染色体優性尿細管間質性腎疾患[ADTKD]、ミトコンドリア病)
前述のアルポート症候群に加え、同じ家系内で腎不全の患者さんが多くいらっしゃる場合に、その家系では腎臓病に関連する遺伝子に原因(変異)がある可能性が高いと考えられます。その中でも遺伝性巣状分節性糸球体硬化症(FSGS)、常染色体優性尿細管間質性腎疾患(ADTKD)、ミトコンドリア病はメジャーな原因となり得ます。原因遺伝子としてはFSGSでは約50種類、ADTKDでは5種類が知られており、症状、治療法や予後予測はそれぞれで異なるがゆえ多岐にわたります。当教室の網羅的パネル遺伝子診断ではこれらの多くの遺伝子を同時にカバーすることで効率的な診断を行なっています。
多嚢胞腎
- 多発性嚢胞腎とは?
-
多発性嚢胞腎は、両側の腎臓に嚢胞(嚢胞液が入った袋)ができ、それらが年齢とともに増えて大きくなっていく遺伝性の病気です。嚢胞が増えて大きくなると、腎機能が低下していきます。成人で見つかる多発性嚢胞腎はほとんどが常染色体優性多発性嚢胞腎(ADPKD)で、腎臓以外の臓器にも特徴的な合併症が生じることがある全身の病気です。最近、多発性嚢胞腎は難病指定されると同時に、進行抑制効果のあるバソプレシン受容体拮抗薬治療薬が新しい治療薬として認可されました。治療薬の適応には、正しい診断と評価が必要であり、腎臓専門医/難病指定医のいる施設での診療が重要になっています。
- 患者さんの経過は?
-
日本のADPKD患者数は約31,000人と推定され、60歳までに約半数の患者さんが、血液透析などの腎臓の代わりの役割を担う治療(腎代替療法)が必要な末期腎不全になります。ADPKDの患者さんの経過は同じ家系内でも個人差が大きいのですが、60歳までに患者さんの約半数が末期腎不全となり、透析療法や腎臓移植が必要となります。
- ADPKD以外の多発性嚢胞腎について
-
親がADPKDの場合、子どもがADPKDになる確率は50%です。しかし、両親に多発性嚢胞腎が認められない成人症例が10%程度いることが知られています。小児科領域においては家族歴を認めない多発性嚢胞腎を起こす疾患がいくつかありますが、成人で発見される家族内発症がない多発性嚢胞腎の場合、臨床の現場ではADPKDとして診療されることが多いのが現実です。しかし、その他の嚢胞性腎疾患の可能性も否定できず、正確な遺伝学的背景は十分に明らかになっていませんでした。実は腎臓に多発性嚢胞腎を起こす疾患はADPKDだけではなく、通常は小児科で見つかる常染色体劣性多発性嚢胞腎やネフロン癆などといった疾患があるのですが、多発性嚢胞腎を起こすすべての原因遺伝子を一度に検査することは、従来の手法では大変な労力を要しました。そこで、我々は次世代シークエンサーを用いて嚢胞性腎疾患の原因となる 70個以上の遺伝子を網羅的に解析できる遺伝子診断パネルを作成しました。特に、家族歴や画像が典型的でない多発性嚢胞腎患者につきましては、この遺伝子診断パネルが有効であることを我々は報告しました(Clin Genet. 2018)。また、この遺伝子診断パネルでは腎生検で常染色体劣性多発性嚢胞腎やネフロン癆などを疑われた症例も網羅的に遺伝子検査できるようになっています。
IgA腎症
IgA腎症は、日本人が最も罹患する慢性糸球体腎炎(腎臓の糸球体に炎症を生じる疾患の総称)で、透析療法や腎移植が必要となってしまう代表的な腎臓病です。疫学調査によるとIgA腎症の患者さんは3万人を超えるとされます。私たちが左右に持つ腎臓にはそれぞれ、糸球体と呼ばれる尿の濾過装置が約100万個存在します。糸球体に免疫グロブリン のIgAという蛋白が沈着し、炎症を起こして蛋白尿や血尿が出ます。典型的には血尿と蛋白尿が続いて学校検尿で発見されることや、風邪をひいた後にコーラのような色の尿や真っ赤な尿(肉眼的血尿)で気づかれることが多いです。
病気の原因は様々で、遺伝的素因やリンパ球の機能異常、細菌やウイルス感染症などによって、異常なIgAが扁桃腺や骨髄で産生されて糸球体に溜まるとされています。成人では20年で30~40%が、小児では15年で10%前後が末期腎不全(透析療法や腎移植が必要な状態)に至ります。そのため、将来の末期腎不全を避けるためには、初期の検尿異常(蛋白尿や血尿)を決してあなどらずに早期に診断をつけること、いくつか確立された治療法によって早期に寛解(蛋白尿と血尿を消失させること)を目指すことが重要です。
まず確定診断のために腎生検と呼ばれる組織診断を行います。しっかりと局所麻酔を行い、超音波で腎臓を見ながら糸球体を含む腎組織を採取します。治療は、先ほどの異常なIgAの産生を抑えるための扁桃摘出術と、糸球体の炎症を抑えるためのステロイドパルス療法を組み合わせた治療(扁摘パルス療法)を行います。IgA腎症が発症してから治療までの期間が短ければ短いほど効果が期待されます。血尿が最初に出現してから3年以内に扁摘パルス療法を行なうと80%以上の寛解率が期待できます。患者さんの重症度に応じ多剤併用療法(カクテル療法:ステロイド、免疫抑制薬、抗血小板薬など)を行うことや、軽症の方では扁摘パルス療法を行わない治療法(アンギオンテンシン変換酵素阻害薬やアンギオテンシン受容体拮抗薬など)も実施します。食事療法としては減塩を基本とし、腎機能の低下を認める場合はたんぱく質制限が必要になることもあります。その他禁煙や、肥満の方は減量が勧められます。
当診療科では年間500~600人の入院患者さん、17000人前後の外来患者さんの診療にあたっています。中でもIgA腎症の患者さんも多く、初診の方や入院しながら腎生検を行っている方・ステロイド治療中の方、寛解後に外来フォロー中の方など、様々な治療フェーズの患者さんの診療にあたっています。扁桃摘出術の有効性は最近本邦からの大規模研究結果で再確認されましたが(JAMA Network Open 2019; 2(5): e194772)、当院では耳鼻咽喉科と連携しながら、最適な治療時期を選んで実施しています。常に最新の知見を生かしながら診療を行っています。