「αミオシン重鎖遺伝子変異は洞不全症候群の原因となる」【木村彰方 教授】FINDING / PRESS
洞不全症候群の分子病因解明に関する長崎大学 蒔田教授らとの共同研究の成果が米国循環器学会誌(Circulation Arrhythmia and Electrophysiology)にオンライン速報版で発表(2015.2.25付)されました。
αミオシン重鎖遺伝子変異は、心筋細胞のサルコメア整合性の障害と電気刺激伝播速度の遅延を来たし、洞不全症候群を引き起こす【共同研究拠点:木村教授】
石川泰輔 助教(長崎大学、東京医科歯科大学)、Chuanchau J. Jou博士(Utah大学)、野上昭彦 教授(筑波大学)、木村彰方 教授(東京医科歯科大学)、蒔田直昌 教授(長崎大学)他
Ishikawa T, Jou CJ, Nogami A, Kowase S, Arrington CB, Barnett SM, Harrell DT, Arimura T, Tsuji Y, *Kimura A, *Makita N: A novel mutation in a-myosin heavy chain gene is associated with sick sinus syndrome. Circ Arrhythm Electrophysiol, (*co-corresponding authors)[Epub ahead of print, Feb.25, 2015]( doi: 10.1161/CIRCEP.114.002534)
成果のポイント
- 洞不全症候群患者にαミオシン重鎖遺伝子(MYH6)変異を見出しました。
- MYH6変異は、ミオシン重鎖と心筋ミオシン結合タンパクCとの結合性を増強しました。
- MYH6変異は、心筋細胞のサルコメア整合性を障害しました。
- MYH6変異は、心房筋細胞株の細胞間電気興奮伝播を遅延させました。
- MYH6変異は、myh6発現欠損ゼブラフィッシュの徐脈を軽減する効果が十分ではありませんでした。
研究の背景
洞不全症候群(sick sinus syndrome, SSS)は、洞房結節の機能異常によって徐脈を来たす比較的頻度の高い不整脈です。臨床的には徐脈以外に失神発作や心不全を生じますが、加齢や心筋虚血が原因と考えられる症例に加えて、原因不明の症例が多く存在します。原因不明の症例の一部には家族歴が認められることがあり、そのような症例の遺伝子解析から、心筋Naチャネル遺伝子(SCN5A)、アンキリンB遺伝子(ANK2)、過分極活性型カチオンチャネル遺伝子(HCN4)の変異がSSSの原因となることが報告されています。しかしながら、それらの遺伝子には変異が見出されない症例も多数存在しており、その他の原因遺伝子があると考えられています。最近、ゲノムワイド関連解析からαミオシン重鎖遺伝子(MYH6)の遺伝子多型A1101Vが心拍数と関連することが判明し、また、アイスランド人においてMYH6の稀なバリアントR721WとSSSとの関連が報告されています。しかし、これらのMYH6変異がもたらす機能変化や変異とSSS病態との関連は不明でした。ヒト心臓では、αミオシン重鎖(MYH6)とβミオシン重鎖(MYH7)が発現していますが、MYH6は胎児期の心臓で主に発現し、生後はMYH7の発現にスイッチすることが知られており、成人の心室筋ではMYH7が主体ですが、心房筋ではMYH6が発現しています。これまでの研究で、MYH7変異が遺伝性心筋症(肥大型心筋症や拡張型心筋症)の原因となることが報告されており、それらのMYH7変異がもたらす収縮機能異常が明らかにされています。一方、MYH6変異は一部の肥大型心筋症の症例に見出されていますが、その機能異常は明らかではありません。さらに、心房中隔欠損症などの先天性心奇形症例でもMYH6変異が見出されていますが、それらのMYH6変異は、サルコメア断裂をもたらすことや、サルコメア形成を促進することで心臓、とりわけ心房の形態形成を障害するのではないかと考えられています。しかし、原因不明のSSSでは、一般に心房の形態異常は観察されません。
本研究の成果
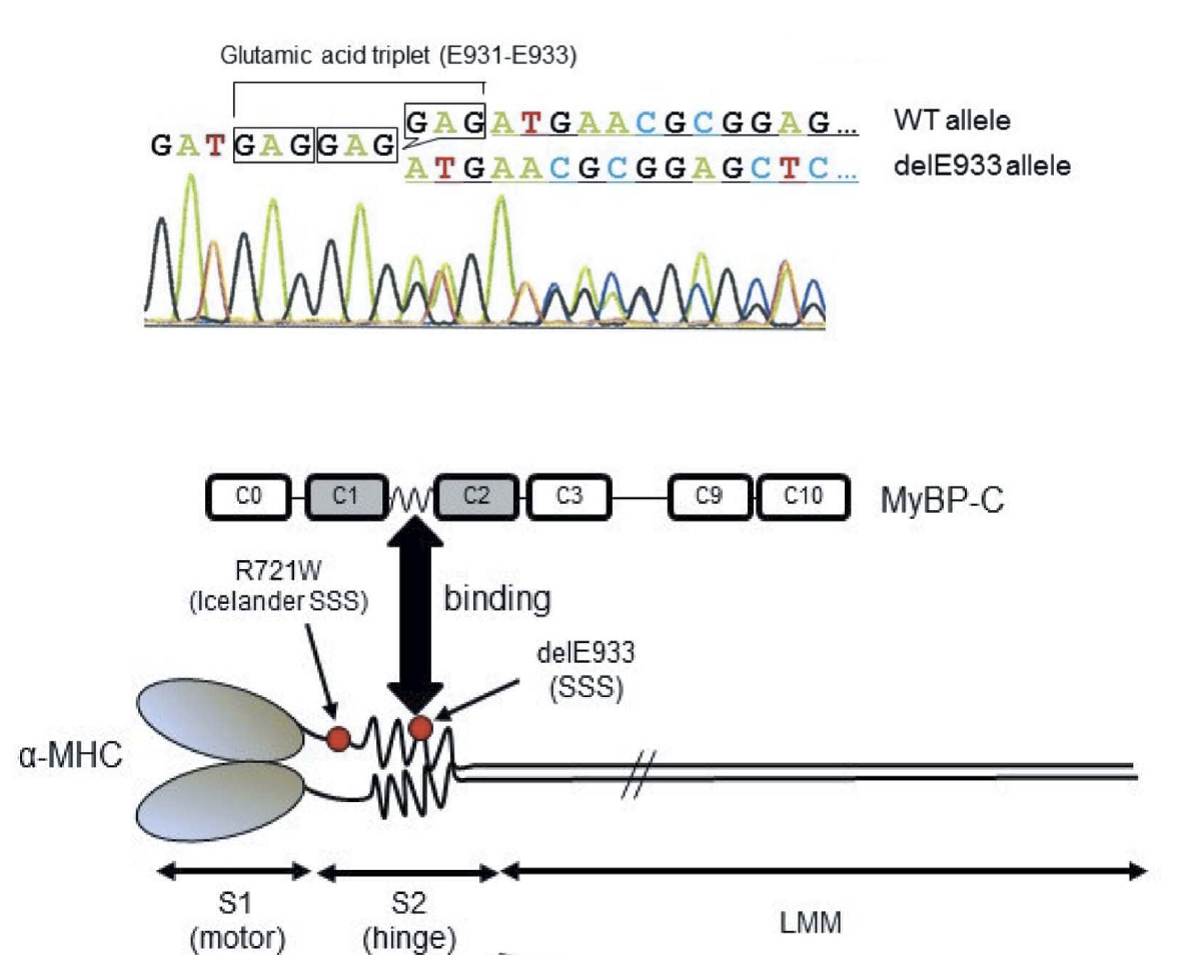
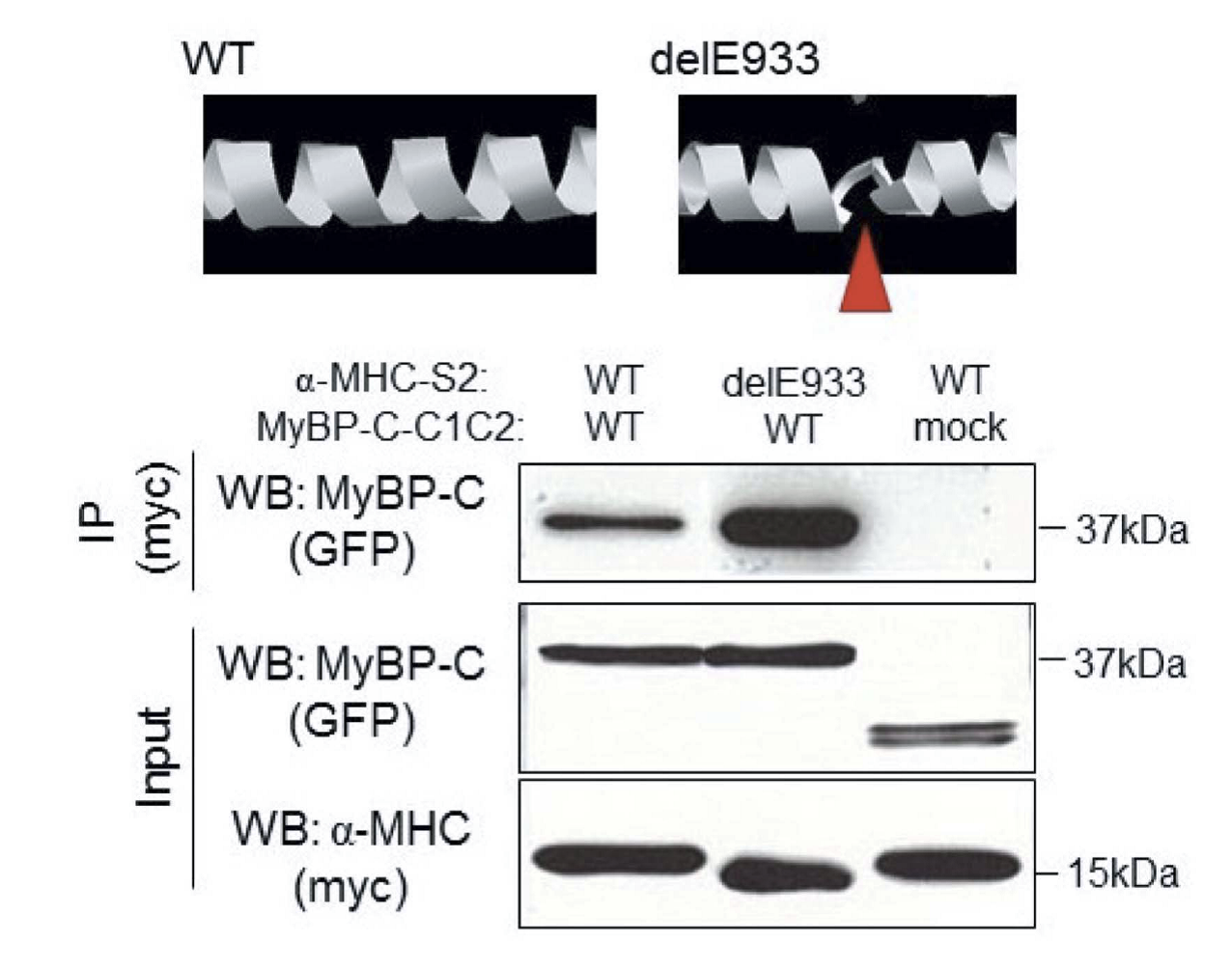
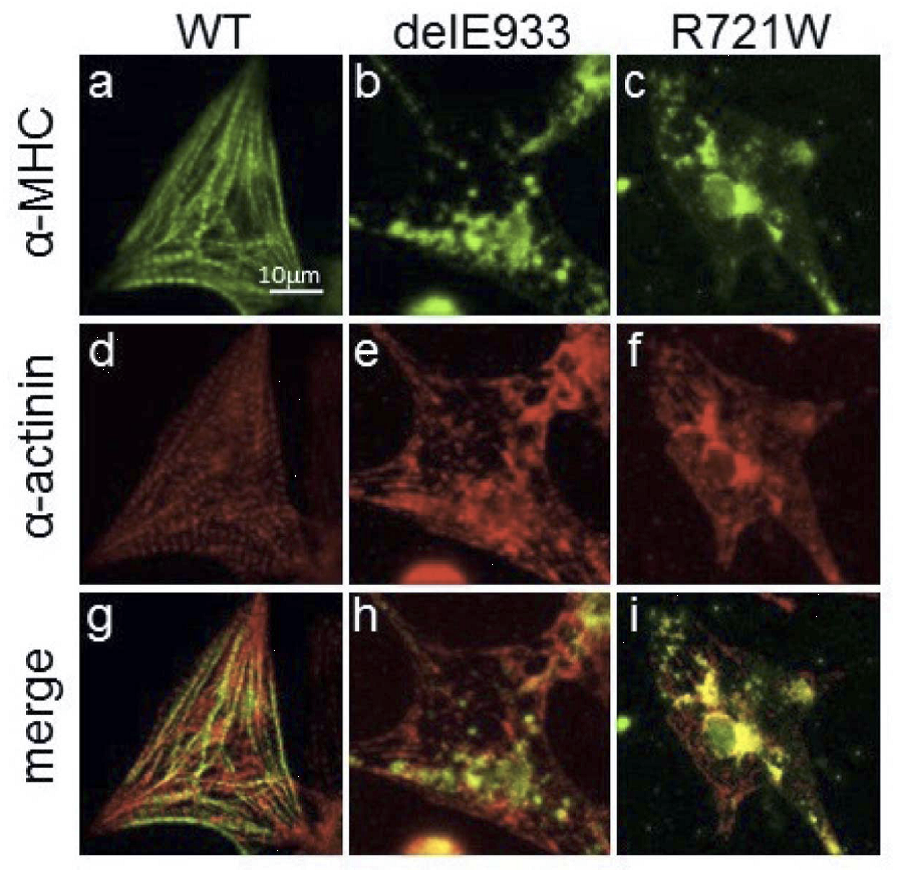
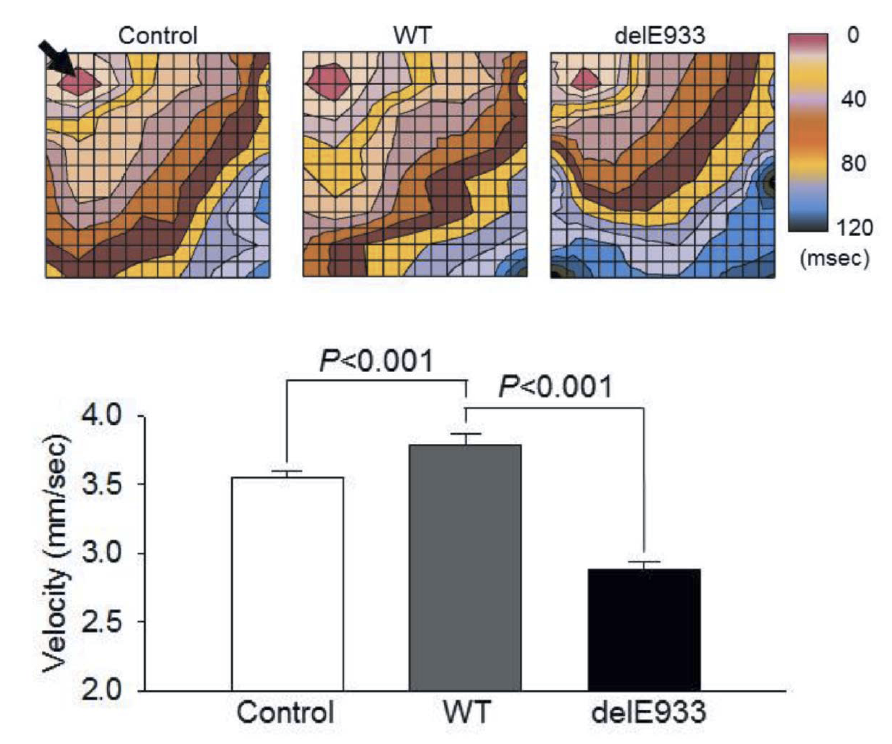
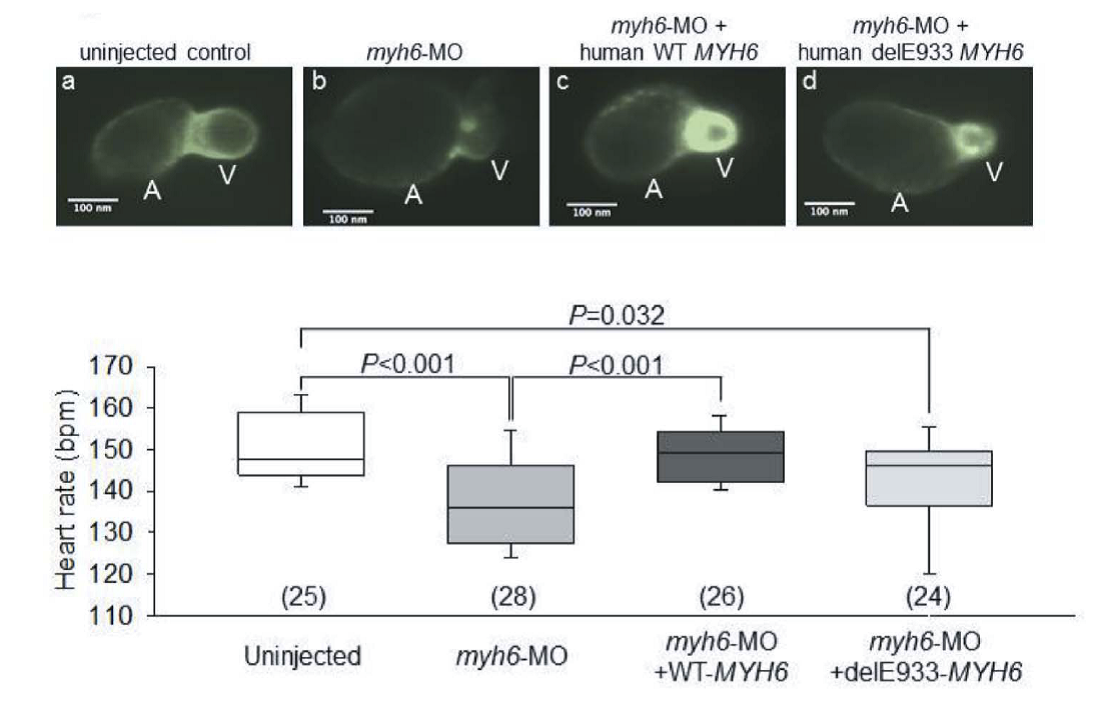
本研究では、SCN5AおよびHCN4に変異が認められない日本人SSS症例9例を対象として、ダイレクトシークエンスによってMYH6変異を検索しました。その結果、1例に第22エクソン中のコドン933番の3塩基欠損(delE933)が検出されました(図1上)。この変異は日本人健常者400名や、国際的な健常者塩基配列データベース(日本人健常者約1,200名を含む)には認められないものでした。また、変異はミオシン重鎖の頚部S2ヒンジ領域でミオシン結合タンパクCとの結合領域に位置するものでした(図1下)。さらに、この症例には、SCN5A、HCN4以外にもSCN3B、KCNJ3、KCNJ5、GJA5などのチャネル遺伝子にも変異がありませんでしたので、delE933変異がSSSの原因であることが示唆されました。ついで、ミオシン重鎖の3Dモデルを構築すると、delE933変異はαヘリクス構造に変化をもたらすと推定されました(図2上)ので、ミオシン結合タンパクCのC1~C2ドメインとの結合性を検討すると、変異があるとこれらの結合性が増強することが判明しました(図2下)。そこで、ラット心筋細胞に正常MYH6、delE933変異MYH6、R721W変異MYH6をそれぞれ導入して検討したところ、delE933、R721W変異はいずれも心筋細胞内で核膜近傍へ集積し、サルコメア構造の崩壊が観察されました(図3)。このことはSSSに関連するMYH6変異はサルコメア整合性の障害をもたらすことを示唆します。ついで、del933E変異による心房筋の電気生理学的機能への影響を検討するために、心房筋由来細胞株HL-1に正常MYH6およびdel933E変異MYH6をそれぞれ導入し、64-well電極上で細胞間電気刺激伝播速度を検討しました。その結果、delE933は伝播遅延を来たすことが判明しました(図4)。さらに、個体レベルでの機能異常を検証することを目的として、ゼブラフィッシュを用いた研究を行いました。まず、モルフォリーノを用いてゼブラフィッシュのMYH6発現を低下させると、心房の拡大と徐脈が生じることを確認しました(図5)。つまり、MYH6は心房筋の収縮機能と洞機能を司ることが示唆されました。ついで、正常MHY6およびdel933E変異MYH6をそれぞれ導入したところ、正常MHY6では徐脈から回復しましたが、del933E変異MYH6による徐脈回復は十分ではありませんでした(図5)。以上のことから、MYH6変異(delE933変異)がSSSの原因となると考えられ、そのメカニズムとして心房筋の分化異常の関与が推定されます。
謝辞
本研究の一部は、難治疾患共同研究拠点共同研究経費(共同研究者:長崎大学医学部・教授 蒔田直昌、平成25年度~)の助成を受けて実施しました。