
Top > 研究内容
![]()
1.遺伝子改変モデルを用いた「新たな慢性アレルギー炎症誘発機構」の解明
〜血球細胞のなかで最も数の少ない好塩基球が、実は慢性アレルギーをひきおこす主役であった〜
2.アレルギー疾患・免疫不全症の責任遺伝子・関連遺伝子の同定ならびに治療に向けた病態解析
〜高IgE症候群の責任遺伝子(STAT3ならびにTyk2)を世界に先駆けて同定し、その病態を明らかにした〜
3.リンパ球の発生機構の理解 〜免疫不全症・自己免疫疾患の発症機構の解明〜
1.遺伝子改変モデル動物を用いた「新たな慢性アレルギー炎症誘発機構」の解明
〜血球細胞のなかで最も数の少ない好塩基球が、実は慢性アレルギーをひきおこす主役であった〜
日本をはじめとする先進諸国ではアトピー性皮膚炎や喘息、花粉症に代表されるアレルギー性疾患が年々増加傾向にあります。現在までのところ有効な根本治療法が見いだされていないため、患者の肉体的、精神的、経済的負担は大きく、深刻な社会問題となっています。新たな創薬の標的を見いだすためには、免疫学的アプローチによってアレルギー性疾患の本態を明らかにする必要があります。花粉症のような即時型アレルギー反応に関してはその発症機序がかなり解明されていますが、アトピー性皮膚炎や喘息にみられるような慢性アレルギー炎症病態に関しては、いまなお未知の部分が数多く残されています。
アレルギー性疾患の病態を解析して新しい治療法を開発するためにはヒトのアレルギー疾患を再現できるような動物モデルの樹立が必須です。そこで私たちは世界に先駆けて、アレルゲン特異的なIgEを構成的に発現するトランスジェニックマウスを樹立しました(Int. Immunol.,1999)。 このモデルマウスの解析から、IgEが即時型アレルギー反応だけではなく慢性アレルギー炎症の病態形成にも深く関わっているという新事実が明らかとなりました (J. Allergy Clin. Immunol., 2003)。この新発見は「IgEが即時型のアレルギー反応をひきおこす」「T細胞が遅延型免疫応答をひきおこす」という既成概念では説明できない、全く新しい慢性アレルギー炎症誘導機構が存在することを意味します。 |
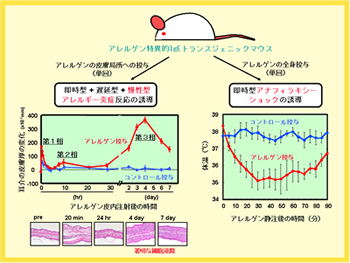 |
 |
そこでさらに、この新規の慢性アレルギー炎症がどのような細胞によってひきおこされるのかを詳細に解析した結果、驚くことに、アレルギー反応をひきおこす主役と考えられているマスト細胞(肥満細胞)やT細胞がまったく存在しない変異マウスにおいても、慢性アレルギー炎症が観察されました。細胞移入実験などにより責任細胞を絞り込んでいき、ついに、血球細胞のなかで最も数が少ない好塩基球が慢性アレルギー炎症をひきこす主役であることをつきとめました(Immunity, 2005)(2005年8月24日付けの朝日新聞、毎日新聞、日経BPなどに記事が掲載されました)。これまで好塩基球の生体内における役割はまったくわかっていませんでしたが、本発見によって、好塩基球がマスト細胞とはちがったユニークな役割を担っていることがはじめて証明されました。 好塩基球の研究を進めていく上で大きな障害となっているのは、好塩基球のみを欠損する動物が存在しないことです。最近私たちの研究室で、マウスの好塩基球を特異的に除去できるモノクローナル抗体の樹立に成功しました(Blood, 2007; J. Immunol., 2007)。この抗体を用いた実験から、上記の慢性アレルギー炎症において、好塩基球はエフェクター細胞(実行部隊)ではなくイニシエーター細胞(指揮官)として機能していることが明らかとなりました(Blood, 2007)。さらに、当初の予想に反して、即時型アレルギー反応である全身性アナフィラキシーにおいても、好塩基球はマスト細胞とは明らかに異なる役割を果たしていることが判明しました(Immunity, 2008 ) (2008年3月14日付けの毎日新聞などに記事が掲載されました)。 現在、好塩基球がどのような分子を介して アナフィラキシー・ショックや 慢性アレルギー炎症をひきおこすのか、その分子機構の解明を進めており、好塩基球やその分泌物質を標的にした新規アレルギー治療法の可能性を追求しています。 |
(1)アレルギー疾患モデル動物(アレルゲン特異的IgEトランスジェニックマウス)の樹立
(2)好塩基球が主役を演じる新たな慢性アレルギー炎症誘発機構の解明と治療への応用
(3)IgEによるIgEレセプターの発現制御機構の解明―創薬の新たなターゲット

2.アレルギー疾患・免疫不全症の責任遺伝子・関連遺伝子の同定ならびに治療に向けた病態解析
〜高IgE症候群の責任遺伝子(STAT3ならびにTyk2)を世界に先駆けて同定し、その病態を明らかにした〜
高 IgE症候群は、免疫不全症(繰り返す肺炎や皮膚膿瘍)でありながら重症のアトピー症状(アトピー性皮膚炎、血中IgE高値)を示す大変ユニークな原発性免疫不全症です。多くの症例は孤発性(家族歴が無く、遺伝性が明確でない)ですが、少数ながら常染色体優性型あるいは常染色体劣性型の遺伝形式を示す家系が報告されています。臨床症状から、1型と2型に大別され、1型は免疫系の異常に加えて骨・歯・軟組織の異常をも伴う多臓器疾患ですが、2型で認められる異常は免疫系に限局しています。弧発型と常染色体優性型の多くは1型で、常染色体劣性型は2型の症状を示します。疾患自体は40年以上前にすでに報告されていますが、これまで両タイプともその原因遺伝子ならびに病気の発症メカニズムはまったく闇の中にありました。 (1) 2型高 IgE症候群の責任遺伝子の同定と病態解析 私たちは、まず、高 IgE血症、アトピー性皮膚炎ならびに細胞内寄生細菌・ウイルスによる感染症を繰り返すという2型高IgE症候群症状を呈する症例の末梢血細胞の各種サイトカインに対する反応性を調べました。その結果、患者細胞では、I型インターフェロン(IFN)、インターロイキン-6(IL-6)、IL-10、 IL-12、IL-23を含む多岐にわたるサイトカインに対する反応性が著しく低下していることを見いだしました。そこで、これらのサイトカイン受容体の下流で機能する各種シグナル伝達分子の遺伝子を解析し、JakファミリーキナーゼのひとつであるTyk2をコードする遺伝子に変異を発見しました。対応する正常遺伝子を患者細胞に発現させたところ、各種サイトカインに対する不応答性は正常化したことから、この遺伝子が高IgE症候群の責任遺伝子であることが証明されました(Immunity, 2006; Curr. Opin. Allergy Clin Immunol. 2007) 。Tyk2ノックアウトマウスに比べると、Tyk2欠損患者におけるサイトカイン・シグナル伝達障害はずっと重篤であり、ヒトではマウスに比べて、Tyk2の重要度が極めて高いことがわかりました。Tyk2欠損患者における多彩な臨床症状は、多岐にわたるサイトカイン・シグナル伝達障害によるものと考えられます。すなわち、繰り返すウイルス感染症は、I型IFNのシグナル伝達障害によるもの、細胞内寄生細菌による感染症はIL-12シグナル伝達障害のためのTh1応答低下によるもの、高IgE血症とアトピー性皮膚炎はIL-10シグナル伝達障害などのためのTh2応答亢進によるものと説明できます。 (2) 1型高 IgE症候群の責任遺伝子の同定と病態解析 ひきつづき、1型高 IgE症候群の原因も複数のサイトカイン・シグナル伝達に関わる分子の異常ではないかという仮説のもと候補遺伝子を絞って解析を進めた結果、ついにSTAT3遺伝子のヘテロ変異を発見しました(Nature, 2007)(2007年8月6日のNHK朝のニュース、朝日新聞、毎日新聞、読売新聞、日本経済新聞、産経新聞などで研究成果が報道されました)。8名の弧発型高IgE症候群患者に認められた変異はすべてSTAT3のDNA結合領域に存在し、変異STAT3ではDNA結合能が失われていました。この変異STAT3は共存する正常STAT3に対してドミナント・ネガティブに作用するため、患者細胞におけるSTAT3のDNA結合能は正常の1/4しかないことがわかりました。STAT3変異は弧発型高IgE症候群患者の両親や兄弟には存在しないことから、先天性でありながら遺伝性のものではなく突然変異によるものであると考えられます。STAT3は30近くのサイトカイン、増殖因子、ホルモンのシグナル伝達に関与しているとの報告があることから、SATA3の変異は高IgE症候群に認められる多岐にわたる臨床症状をよく説明できるものと思われます。 高 IgE症候群は早期診断が困難で、乳幼児・児童期では重症のアトピー性皮膚炎と診断されていたものが、後になって症状が集積してきてはじめて高IgE症候群と診断された症例もたくさんあります。本研究で原因遺伝子(STAT3ならびにTyk2)が同定されたことにより、早期の確定診断(遺伝子診断)が可能となり、将来的な遺伝子治療への道も開けました。高IgE症候群では肺炎にともなう重症合併症が大きな問題となりますが、早期診断を行って、ごく早い段階から感染予防の処置を施すことで、重篤合併症を回避することができます。高IgE症候群に認められる慢性皮膚炎は重症アトピー性皮膚炎と類似していることから、本研究成果が、より一般的なアトピー性皮膚炎の病態解明や新規治療法の開発につながるのではないかと期待して、さらに研究を進めています。 |
3.リンパ球の発生制御機構の理解?免疫不全症・自己免疫疾患の発症機構の解明
代表的な先天性免疫不全症であるBruton型無γグロブリン血症の患者では、免疫系において抗体産生を担うB細胞がほとんどまったく産生されないため、すべてのクラスの抗体ができず、重篤な感染症を繰り返します。1993年にその原因遺伝子として新規チロシンキナーゼBtkをコードする遺伝子が同定されて以来、峯岸ら(現在、本分野の助教授)によってB細胞欠損型先天性免疫不全症の原因遺伝子が次々と同定されました(New Engl. J. Med., 1996; J. Exp. Med., 1998; Proc. Natl. Acad. Sci. U.S.A., 1999; J. Clin. Invest., 1999; Clin. Immunol., 1999; Science, 1999; Immunol. Rev., 2000)。 大変興味あることに、これまでに同定された5つの責任分子(μ鎖、λ5、Igα、Btk、BLNK)はいずれも、B細胞分化の初期段階において選択的に発現するプレB細胞レセプターと関連するもので、プレB細胞レセプターの構成分子あるいはそのシグナル伝達に関与する分子です。すなわち、骨髄におけるB細胞の発生・分化誘導にプレB細胞レセプターが決定的な役割を果たしていることが明らかとなりました。 |
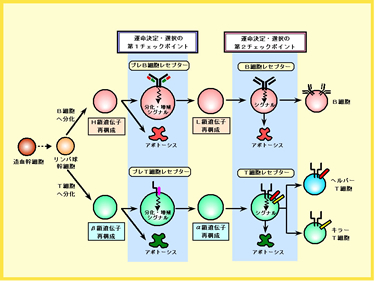 |
私たちは、最近、骨髄や胎児肝臓などB細胞が発生・分化する場において、プレB細胞レセプターを形成できるプレB細胞と形成できないプレB細胞とを見分けて別個に単離する方法を確立しました。この方法を応用してプレB細胞の解析を進めた結果、プレB細胞レセプターが免疫グロブリンH鎖の品質を厳密にチェックして、最終的なB細胞のレパートリー形成に大いに貢献していることが明らかとなりました(J. Immunol., 2006 )。 また、胎児肝臓でプレB細胞レセプターによる選別を受けたユニークなH鎖を発現したB細胞が、成体になっても脾臓の辺縁帯と呼ばれる特殊な領域で保持され続けていることを見いだしました(Int. Immunol., 2005)。 このようなプレB細胞レセプターによるH鎖の品質管理が破綻すると、免疫不全症や自己免疫疾患が発症する可能性が考えられ、現在、モデル動物を作製して品質管理破綻による免疫異常症の発症メカニズムを解明しています。 |
|
(1)プレB細胞レセプターによる免疫グロブリンの品質管理とB細胞レパートリーの形成制御
(2)異常μ鎖(軽鎖非依存性μ鎖)によるB細胞分化障害〜免疫不全症・自己免疫疾患の発症
