2-3-10 Kanda-Surugadai Chiyoda-ku Tokyo 101-0062, JAPAN
Institute of Science Tokyo, Institute of Integrated Research
Advanced Research Initiative, Pathological Cell Biology
TEL: 03-5280-8082 FAX: 03-5280-8079
Our Subject
Our laboratory is engaged in research centered on three core themes: (1) elucidation of the physiological and pathological roles of Golgi-membrane-associated degradation (GOMED), a cellular function we originally identified; (2) mechanistic dissection of cell death pathways and the development of therapeutic interventions for diseases caused by their dysregulation; and (3) investigation of disease pathogenesis associated with mitochondrial and Golgi apparatus dysfunction. Through these lines of inquiry, we ultimately seek to advance a fundamental understanding of the operational principles that underlie cellular life.Main Research Themes
1. Characterization of a Novel Protein Degradation Pathway: Golgi-Membrane-Associated Degradation (GOMED)
- A, Dissection of the molecular mechanisms underlying GOMED
- B, Elucidation of the physiological significance of GOMED in vivo
- C, Application of GOMED insights to disease contexts
2. Investigation of Cell Death Mechanisms
- A, Molecular analysis of apoptotic pathways
- B, Characterization of non-apoptotic cell death mechanisms, including autophagic cell death and necrosis
- C, Exploration of the biological relevance of cell death in vivo
- D, Development of therapeutic approaches targeting cell death-related pathologies
3. Analysis of Organelle Membrane Dynamics
- A, Investigation of the interplay between Golgi membrane morphology and its functional regulation
- B, Pathophysiological analysis of diseases caused by mitochondrial dysfunction
1, Molecular Mechanisms and Physiological Functions of GOMED
Golgi-membrane-associated degradation (GOMED) is a recently identified protein degradation pathway executed via structures that resemble those of autophagy. However, it differs substantially from canonical autophagy in terms of its inducing stimuli, effector molecules, and substrate specificity. While autophagy is mediated by membranes derived from the endoplasmic reticulum, GOMED uniquely utilizes membranes from the Golgi apparatus (Figure 1).
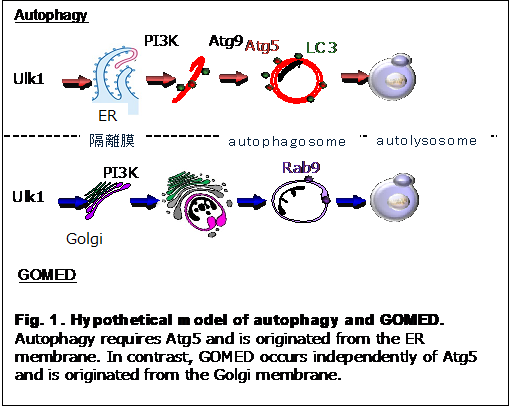
Under physiological conditions, the cis-, medial-, and trans-Golgi cisternae function as an integrated unit to properly modify and deliver secretory and membrane proteins. However, when secretory protein trafficking becomes excessive or protein abnormalities arise, GOMED is activated. During this process, portions of the trans-Golgi network dissociate from the medial cisternae, become spherical, and encapsulate aberrant proteins. These encapsulated proteins are then degraded by lysosomal enzymes.
Although GOMED and autophagy coexist within the same cell, their activation and substrate specificity differ markedly depending on the nature of the stimulus and the proteins to be degraded. Consequently, their physiological roles are distinct and non-overlapping.
The molecular machinery of GOMED is highly conserved from yeast to mammalian cells. In the initiation phase, key autophagy-related proteins such as Ulk1 and Beclin 1 are involved, followed by the downstream activation of Wipi3. Upon GOMED induction, Wipi3 translocates from the cytoplasm to the Golgi apparatus, where it contributes to membrane remodeling. The curved Golgi-derived membranes enwrap specific proteins from both the cytosol and the Golgi lumen, which are ultimately degraded in lysosomes.
A major distinguishing feature of GOMED, compared to conventional autophagy, lies in the identity of its target substrates. While autophagy primarily degrades cytoplasmic components such as p62 and LC3, GOMED specifically targets proteins trafficked through the Golgi apparatus.
One prominent example of GOMED function is its role in regulating insulin secretion. In pancreatic β-cells, insulin is synthesized and secreted via the Golgi apparatus. When extracellular glucose levels drop, GOMED is induced to reduce intracellular insulin retention, thereby attenuating further insulin secretion and preventing hypoglycemia.
GOMED also plays a critical role in maintaining neuronal homeostasis. This was demonstrated in mice with neuron-specific deletion of the Wipi3 gene, which exhibited Purkinje cell degeneration and behavioral abnormalities. In this context, GOMED was found to mediate the constitutive degradation of ceruloplasmin, an iron-transport protein. When GOMED is impaired, ceruloplasmin accumulates, leading to iron deposition and the development of neurodegenerative diseases associated with iron overload.
2, Investigation of Cell Death Mechanisms
A large proportion of cells in the human body harbor intrinsic suicide machinery and are capable of initiating self-destruction under appropriate conditions. These programmed cell death pathways function in concert with cellular proliferation and division, contributing to organismal development and tissue homeostasis. Historically, apoptosis was regarded as the sole form of physiological cell death. However, subsequent studies—including our own—have demonstrated the existence of multiple non-apoptotic cell death pathways. It is now recognized that a variety of cell death mechanisms are selectively activated under different physiological and pathological contexts.
Our research aims to:
(1) Elucidate the molecular mechanisms of distinct cell death pathways;
(2) Define the biological significance of these pathways in vivo; and
(3) Explore their implications for the pathogenesis and treatment of human diseases.
The core molecular framework of apoptosis has been extensively characterized. Apoptotic signals often converge at the mitochondria, where they trigger increased membrane permeability. Several pro-apoptotic factors reside in the mitochondrial intermembrane space and are released into the cytosol upon membrane permeabilization. This release leads to the activation of caspases, a family of cysteine proteases that execute the apoptotic program.
Central to the regulation of this process is the Bcl-2 family of proteins, which govern mitochondrial membrane integrity and thus determine cell fate (Nature, 1995; Nature, 1999; Cell, 2003). Nevertheless, several aspects of mitochondrial signaling and its upstream regulation remain incompletely understood. Our laboratory is focused on dissecting these unresolved mechanisms at the molecular level.
B. Molecular Mechanisms of Non-Apoptotic Cell Death
Autophagic Cell Death
We were the first to report that in apoptosis-resistant cell lines—specifically those lacking Bax and Bak—treatment with apoptosis-inducing agents such as chemotherapeutic drugs activates an alternative form of cell death dependent on autophagy (Nature Cell Biology, 2004) (Figure 2). This process, termed autophagic cell death, is now considered to play roles in both physiological and pathological contexts.
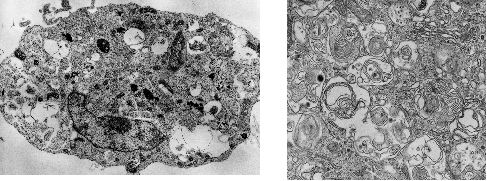
Autophagic cell death is distinct from survival-promoting autophagy and involves excessive or dysregulated autophagic activity that ultimately leads to cell demise. We are currently engaged in detailed molecular characterization of this pathway to clarify its regulatory networks, functional roles in vivo, and potential contribution to disease processes.
C. Elucidating the In Vivo Roles of Cell Death
We have established a comprehensive collection of genetically modified mouse models targeting key regulators of apoptosis, autophagic cell death, and necrosis. Using these models, we aim to dissect the physiological and pathological significance of each cell death modality in vivo. These investigations provide insights into how programmed cell death contributes to tissue homeostasis, development, and disease progression.
D. Application of Cell Death Research to Disease Pathogenesis and Therapy
Aberrant regulation of cell death has been implicated in the onset and progression of a broad spectrum of diseases, including cancer, ischemic injury, and neurodegenerative disorders. Through mechanistic analysis of cell death pathways, we aim to translate our findings into novel therapeutic strategies.
Ischemia–Reperfusion Injury
Our studies using Cyclophilin D-deficient mice have demonstrated marked resistance to ischemia–reperfusion injury in both cardiac and cerebral tissues, implicating permeability transition (PT)-dependent necrosis in the pathogenesis of this condition (Nature, 2005). Building on these findings, we are currently pursuing the development of PT-modulating therapeutic agents.
Radiation-Induced Enteritis
Radiation-induced intestinal damage is primarily driven by apoptosis of intestinal epithelial cells. We previously developed Tat-BH4, a synthetic anti-apoptotic peptide, and demonstrated its efficacy in ameliorating radiation-induced enteritis (PNAS, 2000).
To further advance therapeutic applications, we are currently establishing a high-throughput screening platform for small molecules that modulate mitochondrial membrane permeability, with the goal of identifying novel regulators of apoptosis and necrosis.
3. Analysis of Organelle Membrane Dynamics
A. Functional Correlation Between Golgi Membrane Morphology and Organelle Function
The Golgi apparatus typically operates as an integrated structure composed of cis-, medial-, and trans-cisternae, which collaboratively modify and direct the trafficking of secretory and membrane proteins to their appropriate destinations. However, during the execution of GOMED, portions of the trans-Golgi network become detached from the medial cisternae and undergo vesiculation. In addition, the Golgi apparatus undergoes extensive morphological reorganization during the cell cycle, particularly during mitosis. We are investigating how these structural transitions are coordinated with functional outputs of the Golgi apparatus.
B. Pathophysiological Analysis of Mitochondrial Dysfunction
Mitochondria play a central role in cellular metabolism, notably in ATP production and the tricarboxylic acid (TCA) cycle. At the same time, they serve as a critical regulatory hub for cell death pathways such as apoptosis and necrosis. In both contexts, bidirectional communication between mitochondria and the cytosol is essential for maintaining cellular homeostasis.
Our research focuses on the mitochondrial membrane as a key platform for this inter-organelle communication. We aim to systematically elucidate the structure–function relationships of mitochondrial membrane proteins, including both metabolite transporters and regulators of membrane permeability involved in cell death.
(1) Structural and Functional Analysis of Mitochondrial Membrane Proteins
We are conducting a comprehensive analysis of mitochondrial membrane proteins, encompassing both those that regulate membrane permeability during apoptosis and necrosis, and those that mediate metabolite exchange critical for cellular metabolism.
(2) Therapeutic Targeting of Mitochondrial Dysfunction in Disease
Mitochondrial dysfunction is increasingly recognized as a key pathogenic factor in various diseases, particularly neurodegenerative disorders. In some forms of neurodegeneration, mitochondrial DNA point mutations and resultant oxidative stress contribute to disease progression. Furthermore, several familial neurodegenerative diseases, including certain forms of Parkinson’s disease, are directly linked to mutations in genes encoding mitochondrial proteins. We seek to elucidate the mechanistic link between mitochondrial abnormalities and disease pathogenesis, with the ultimate goal of developing targeted therapeutic interventions.