1)変性における核機能異常の網羅的解析
JSTさきがけの支援を受けて、2003-2006年まで、ポリグルタミン病の核機能異常について、トランスクリプトーム/プロテオームの網羅的解析を行ってきました。この実験には、アデノウィルスベクターを用いた初代培養神経細胞でのポリグルタミンタンパク発現系(前助手の田川さんを中心に開発したもの)(Tagawa et al., Journal of Neurochemistry 2004)を主に用いました。3年以上にわたる苦闘の末、神経変性に関わる新しい分子群(HMGB1/2)を同定し、ショウジョウバエモデルでその病的機能を確認しました。簡単にいうと、DNA高次構造やDNAのヒストンからの解きほぐしを制御するHMGBタンパク質がポリグルタミン病態で神経細胞の核の中で低下すると、核機能の広範な障害が起こり、なかでもDNAの損傷修復に異常が起きるというものです。そして、ショウジョウバエを使った神経変性モデルにおいて、HMGBに治療効果が確認されました。これらの成果については、Nature Cell Biologyに2007年に公表しました。詳細は2007年の所感(移動)およびプレスリリースをご覧ください(リンク)。
さらに、インタラクトーム解析(タンパク質相互の結合関係の網羅的情報解析)の結果から、別なDNA修復タンパク質Ku70が、病態関連タンパクの候補として挙がって来ました。私たちは、Ku70の病態機能を、生化学、細胞生物学、マウスモデルを多角的に用いて検討した結果、ハンチントン病の異常タンパク質がKu70と結合して機能を阻害し、その結果、DNA損傷修復が低下していることを証明しました。そして、Ku70のレベルを少し上げてやるだけで、従来の報告を上回る治療効果が得られることも示しました(Enokido, Tamura, Ito et al., Journal of Cell Biology 2010)。プレスリリースを見る(リンク)。
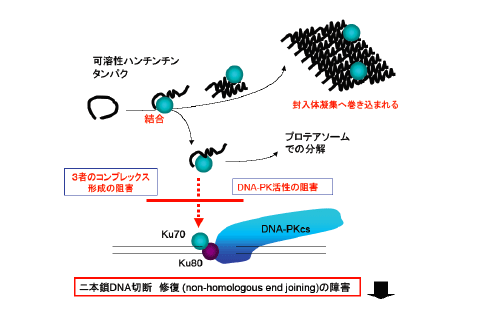
2)転写障害によって誘導される新しい神経細胞死
神経変性疾患の中でポリグルタミン病は核内部に異常タンパク蓄積がみられるという特徴を持っています。これまでに、多数のグループが種々の核タンパク(転写関連因子)と異常ポリグルタミンタンパクの結合を示してきました。私たちの報告したPQBP-1もその一つです。さらに、私たちは異常ポリグルタミンタンパクの発現はBrUの取り込みで解析すると総転写量を減少させること確認しました(Hoshino et al., BBRC 2004)。これらの事実はポリグルタミン病で転写障害が生じていること、さらに転写障害が神経変性へ関与することを強く示唆するものです。そこで、わたしたちは、異常タンパクによって核機能が全体としてどのように変化するのか、またどのようなメカニズムで細胞の変調と細胞死が起きるのかに興味を持って研究を進めてきました.この成果の一つとして、2006年2月のJournal of Cell Biologyに、転写障害によって、神経細胞はきわめて緩慢な非典型的細胞死を起こすことを報告しました(プレスリリースにリンクする)。この細胞死はアポトーシスとは異なり、ネクローシスでもありません。現在は、この細胞死が如何なる核由来のシグナルによるものか、このような細胞死は他の神経疾患でも生じうるのか、という疑問を中心に解析を進めています。
3)ポリグルタミン病と精神遅滞に関与する分子PQBP-1
ポリグルタミン病は遺伝子のエクソン内のCAGリピート配列が異常な 長さに伸長するために起きる一連の遺伝性神経変性疾患群の総称です。遺伝性脊髄小脳変性症、ハンチントン病、球脊髄性筋萎縮症など少なくとも9種類の変性疾患がこの範疇に含まれることが分かっています。ポリグルタミン配列は多くの核タンパクに含まれること、いくつかの疾患原因タンパクの機能が核の生理機能に深く関わっていること、疾患タンパクに結合するもののなかに転写関連因子が多いことなどが明らかにされて、核機能障害がポリグルタミン病の発症につながるとの考え方が有力になってきています。
わたしたちはポリグルタミン配列と結合する新しい分子(PQBP-1, PQBP1)を発見し(Waragai et al., Human Molecular Genetics 1999)、PQBP-1が転写とRNA修飾に関連した機能を果たすことを明らかにしてきました。PQBP-1はポリグルタミン病原因タンパクである変異型のAtaxin-1やhuntingtinと結合した結果、PQBP-1が関与する転写障害やスプライシングの異常を起こすことが想定されます(Okazawa et al., Neuron 2002)。PQBP-1はスプライシングが行われる核のnuclear bodyに存在すること(Okazawa et al., Neuron 2002)や、スプライシング因子U5-15kDと結合すること(Waragai et al., BBRC 2002)もその予想を支持しています。
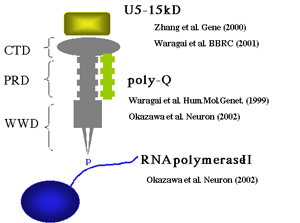
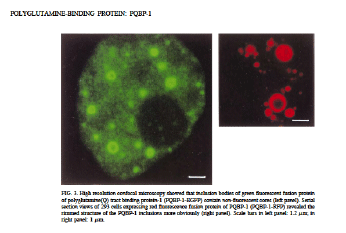
(左図)PQBP-1の結合関係を示す。転写基本因子RNA polymerase IIとRNA splicing factor U5-15kDをリン酸化依存的に結合する。結合には、それぞれWWドメイン(WWD)とC-terminalドメイン(CTD)が使われる。したがってPQBP-1は転写とスプライシングの機能連関に関わる分子であることが予想され、PRドメイン(PRD)に結合する異常ポリグルタミンはこの両機能に悪影響を与えることが予想される。一方、ヒトPQBP1遺伝子に変異が起きて、C-terminalドメインが失われると精神遅滞が生じることが明らかになりました。PQBP1は神経幹細胞にも多く発現しているようであり、現在その機能を解析しています。
(右図)PQBP-1の核内部での分布を示す。中抜けを持ったnuclear bodyを多数形成していることが分かる。nuclear matrixにも薄く分布している。左(緑)の黒く大きく抜けた場所は核小体である。
PQBP-1の発現量はかなり厳密に調節されているらしく、その変化は神経細胞の機能失調や細胞死につながるようです。実際にPQBP-1過剰発現マウスでは運動ニューロンの遅発性神経変性が起きることを発見しました(Okuda et al., Human Molecular Genetics 2003)。さらにこのマウスの運動ニューロン変性過程をマイクロアレイで解析を進めた結果、神経細胞においてミトコンドリアの異常が起きていることがわかりました(Marubuchi et al., Journal of Neurochemistry 2005)。ALS(筋萎縮性側索硬化症)など運動ニューロン疾患では、他のグループからもミトコンドリア異常が報告されており、興味深い結果と考えています。こららの点からPQBP-1を介した神経細胞の変化は変性のモデルと考えられます。
PQBP-1(PQBP1)について新しい情報が2003年にヨーロッパからもたらされました。ヒトPQBP1遺伝子に変異がおこると小頭症を伴う精神遅滞が起きると言う事実です(Kalcheuer et al., Nature Genetics 2003)。PQBP-1が神経変性のみならず、精神遅滞にも関与することになりました。この分子病態的意味については、まだ公表できませんが、クリアーかつエキサイティングな説明が可能です。
一方、PQBP1ノックダウンマウスを作成したところ、興味深い症状を示しました。すなわち、学習の中で不安に関連したものが十分達成できないとのものでした(Ito et al., Hum Mol Genet 2009)。また、ショウジョウバエのPQBP1変異モデルでも学習障害が認められました(Tamura et al., J Neurosci)。しかし、驚いたことに記憶自体には何の問題もありませんでした。マウスとショウジョウバエの双方でシナプス伝達に重要な機能を果たすNMDA受容体のNR1サブユニットの発現低下があり、PQBP1の遺伝子発現機能障害の結果起きていると考えられます。NR1の低下は認知、学習の障害と関連するものと予想しています。さらに、これらのマウスとショウジョウバエの症状はPBAというHDAC阻害剤により、成体になっても改善することも分かりました。つまり、発達障害の症状は固定したものではなく、大人になってからでも薬によって改善する可能性がある、ということが示された訳です。これは、患者さんの治療に大きな光明を与える成果であると考えています。
近年、PQBP1変異の患者さんの報告が相次ぎ、発達障害・精神遅滞においては、fragile-X症候群、Rett症候群に次ぐ、主要な疾患であると考えられています。
4)アルツハイマー病発症の分子メカニズムの研究
アルツハイマー病では、アミロイド前駆タンパク(APP)よりアミロイドが切り出され細胞外に沈着して神経細胞死を起こすというアミロイド仮説が有力です。最近では前駆タンパクの切断に関わる分子複合体の内容が明らかにされ、またアミロイド免疫療法も動物実験で効果が報告され、臨床試験がすでに行われています(現時点では、重篤な白質脳炎を起こす患者が報告され暗礁に乗りあげていますが)。
わたしたちはむしろAPPの生理的機能や核での転写機能に注目し、これまでにアルツハイマー病遺伝子とc-Junの関わりや 人のアルツハイマー病脳の神経細胞においてJNK (c-Junを活性化するアミノ末端部分のリン酸化酵素)が活性化していることを世界に先駆けて報告してきました。 今日ではAPPは現在核伝達シグナルや軸索輸送に関わる分子と考えられています。わたしたちは今後もアルツハイマー病をオリジナルな視点から研究していきたいと考えています。
5)神経幹細胞の分化メカニズムの研究
わたしたちは神経幹細胞についても研究を行っています。わたしたちは20年ちかく前に未分化細胞特異的転写因子Oct-3(Oct-3/4, Oct-4と同じもの)を発見しました(東京大学第一生化学教室<村松正実教授>における濱田博司助教授グループの業績)。岡澤が学位研究として初めて行ったのがOct-3/4のクローニングであり、自らの手でcDNAと染色体クローンの両者を発見しました(Cell 1990; EMBO J 1991)。
研究の発想は、未分化状態のみに発現する転写因子が分化のスイッチになるだろうという予測でしたが、これは多くの研究者が分化後すぐに発現する転写因子を探していたのと逆の発想でした。スイッチ分子かどうかは、その後Oct-3/4のstable cell lineを用いた研究で、Oct-3/4発現と形態変化の相関を確認しましたが、完全な証明には至りませんでした(EMBO J 1993)。しかし、10年ほど経ってから、英国のSmith教授あるいはその共同研究者のNiwa博士らによって、私たちが妄想していたとおりにOct-3/4が胚性幹細胞(ES細胞)の分化のマスタースイッチとして機能することが分かってきました(Cell 1998, Nat Genet 2000)。さらに、そのような知見と関連して、Yamanaka教授らの研究から、Oct-3/4を4転写因子でiPS細胞が創出されました。その後、4因子の中でも、Oct-3/4が最重要であることが多くの論文から示唆されています。例えば、Oct-3/4単独でもiPS細胞が神経幹細胞から作成出来ることがマックスプランク研究所長のHans Schoeler博士らの研究から示されています(Nature 2010)。これらの、国内外で行われたすばらしい研究によってES細胞あるいはiPS細胞におけるOct-3/4の機能が証明されてきました。現在、わたくしたちは神経変性疾患の研究をする立場から神経幹細胞でのOct-3の機能を調べています。このことはやがて神経変性疾患の治療にも役立つ知識を与えてくれるものと考えています。
特に、最近私たちは神経幹細胞においてもOct-3/4が発現していることを発見しました(Okuda et al., Mol Brain Res 2004)。神経幹細胞においても、ES細胞同様にある種の分化のゲートキーパーとして働きうるのかということに興味を持っています。一方、神経幹細胞特異的にOct-3/4を欠失するconditional KOマウスを作製しても、なんら症状が起きない、との報告が最近なされ(Lengner et al., Cell Stem Cell 2007)、Oct-3/4が神経幹細胞に存在するのか?あるいは、以前に報告されたものはpseudogeneではないのか?という論争が起きています。そこで、私たちは精製した神経幹細胞からRNAを抽出し、RT-PCR産物(ゲノムDNAからは増幅されないようにして得られたもの)を直接シーケンスしてみました(Chin et al., BBRC 2009)。その結果、神経幹細胞も明らかにOct-3/4を発現しているとの結論が得られました。したがって、神経幹細胞におけるOct-3/4の機能については更なる検討が必要と考えられます。

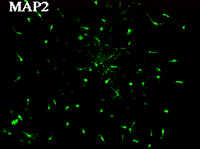
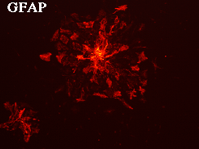
神経幹細胞(neurosphere,上段左)をpoly-L-lysineでcoatingした培養皿に移すと(上段右)、MAP2陽性の神経細胞(下段左)とGFAP陽性のグリア細胞(下段右)に分化する。上段右、下段左、下段右は同一視野。