分子腫瘍医学分野
概要
分化型胃がんや大腸がんなどでは、その原因の解明や予防方法の確立、分子標的治療薬の開発により予後が大きく改善されてきている。一方、肝がん、膵がん、スキルス胃がんなどは依然として予後不良であり、さらなる研究開発が必須である。我々は、難治性がんの分子メカニズムを臨床検体や動物モデルを利用することによりin vivoを反映した実験系で解析を進めており、基礎研究だけに留まらず、常に臨床応用を見据え、がんの新規治療方法を探索している。
プロジェクト
1.難治性がん(肝癌、膵癌、スキルス胃癌など)の分子メカニズム解析
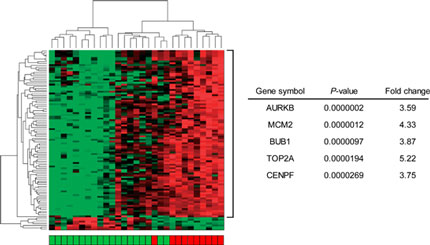
1.1. 肝がんのサブタイプ分類
肝がん臨床検体200例以上のトランスクリプトーム解析を行い、致死的再発、多中心性再発、肉眼型、脈管侵襲、画像所見など臨床病理学的因子と相関する遺伝子群を解析して個別化医療の開発を行っている (Tanaka et al. Hepatology 2011; Sato, Tanaka et al. Hepatology 2013)。 例えば、肝がんの致死的再発は染色体分配を制御するaurora kinase B発現と相関し、多変量解析により唯一の独立規定因子であること、CGH解析によりゲノム不安定性と有意に関連することを明らかにした(Tanaka et al. Br J Surg 2008)。現在、トランスクリプトーム解析に加え、エクソーム解析、メチローム解析などを統合し、肝がんのサブタイプ分類を進めている。
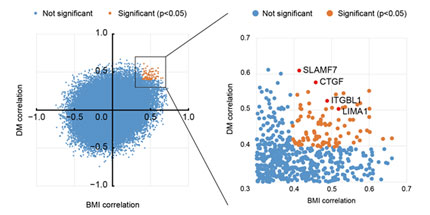
1.2. メタボリック肝がんの解明
近年、B型肝炎ウイルス陰性かつC型肝炎ウイルス陰性の(NBNC)肝がんが増加してきており、糖尿病や肥満などの生活習慣病と関連が強いことが知られてきている。我々はNBNC肝がんの非がん部臨床検体のトランスクリプトーム解析を行い、糖尿病と肥満の両方に相関がある遺伝子としてconnective tissue growth factor(CTGF)を同定した。従来CTGFは肝線維化に寄与していると考えられてきたが、本研究ではマクロファージを誘導することにより腫瘍形成を促進していることを明らかにした(Akahoshi, Tanaka et al. J Gastroentel 2015)。現在、本学分子内分泌代謝学で開発された肥満マウスNASH肝がんモデル (MC4Rノックアウトマウスモデル)とヒト臨床検体の共通メカニズムを共同研究によって解析し、新規診断および治療法開発を進めている。
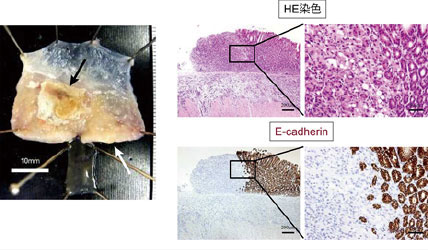
1.3.スキルス胃がんマウスモデルの作製
我々はスキルス胃がんで高頻度に遺伝子異常が認められる細胞接着分子E-cadherinとがん抑制因子p53が胃で特異的にノックアウトされるDCKOマウスを作製した。DCKOマウスではスキルス胃がんが認められ、病理学的にも分子生物学的にもヒトスキルス胃がんをよく模倣していることがわかった(Shimada et al. Gut 2012)。我々は、DCKOマウスの胃がんの初代培養にも成功しており、その細胞株を用いて既知化合物スクリーニングを行い、マウスのみならずヒトのスキルス胃がん細胞にも有効性を示す薬剤を同定している。
2.難治性がんの新規分子標的治療の開発
2.1.aurora kinase B阻害剤
上記の結果を踏まえて、肝がんに対するaurora kinase B阻害剤の有効性を解析している。ヒト肝がん細胞株に対するaurora kinase B阻害剤投与によって、polyploidyを伴うmitotic catastropheが誘導されることを確認した。肝腫瘍モデルを作成してaurora kinase B阻害剤を投与した結果、高い腫瘍抑制効果が認められた(Aihara, Tanaka et al. J Hepatol 2010)。さらにaurora kinase B阻害剤によって誘導されたpolyploid細胞は アポトーシス抑制因子Bcl-xLを高発現することを見出し、aurora kinase B阻害剤投与後にBcl-xL阻害剤を投与すると顕著な相乗的併用効果を示すことを見出した(Matsunaga, Tanaka et al. Ann Surg Oncol 2015)。
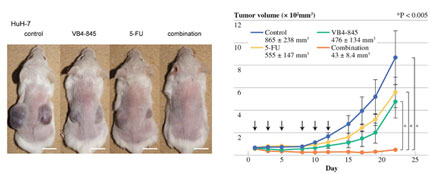
2.2.EpCAM標的治療
肝がん肉眼型の中で多結節融合型は最も予後不良であるが、そのバイオマーカーとしてEpCAMを同定した(Murakata, Tanaka et al. Ann Surg 2010)。 EpCAMは未分化肝芽細胞やオーバル細胞などに発現し,肝がん幹細胞や膵がん幹細胞の特異的マーカーとしても報告されている。抗EpCAM単鎖抗体とエクソトキシンを融合した抗体剤を用いた検証により、肝がん細胞の幹細胞性を抑制し、腫瘍抑制が認められた (Ogawa, Tanaka et al. Ann Surg Oncol, 2014)。
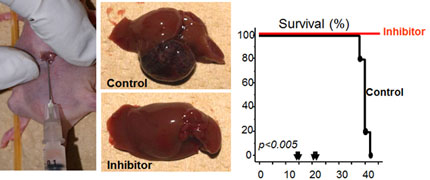
2.3.血管新生阻害剤
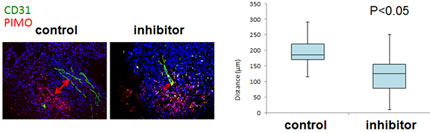
肝がんは典型的な血管新生を示す腫瘍であり、以前より我々は肝がん血管新生に対する標的分子を解析している (Tanaka et al. J Clin Invest 1999, Cancer Res 2001, Hepatology 2002, J Hepatol 2006, Cancer Sci 2009, Semin Oncol 2012)。 肝腫瘍モデルを用いた解析によって、VEGFR/ aurora血管新生阻害剤の抗腫瘍効果が、腫瘍内低酸素領域の低下と有意に相関することを証明した( Nakao, Tanaka et al. Cancer Sci 2015)。現在、血管新生阻害剤の臨床バイオマーカーを探索し、その制御メカニズム解明を進めている。
3.難治性がんのエピゲノム変化を指標とした診断・治療の臨床応用
SET7/9
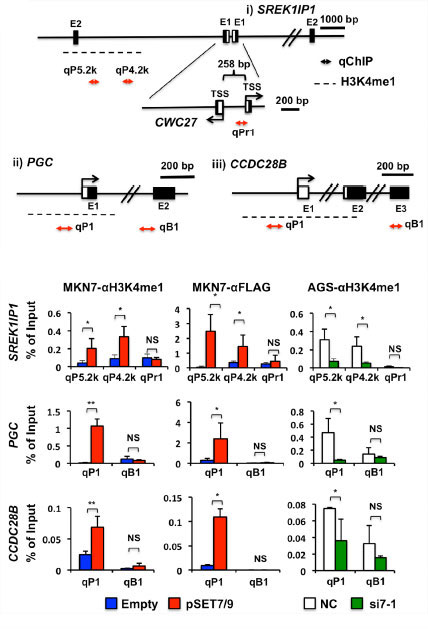
ヒストンメチル化酵素SET7/9は、ヒストンH3の4番目のリジンモノメチル化による遺伝子発現の活性化と非ヒストンタンパク質のリジンメチル化による翻訳後修飾の2つの機能を持っている。我々は約30%の胃がん組織でSET7/9発現が低下していることを明らかにした。胃がん細胞株でSET7/9をノックダウンすると、細胞増殖、遊走能および浸潤能が有意に亢進した。マイクロアレイやChIP解析により、SET7/9はSREK1IP1やPepsinogen C(PGC)遺伝子のヒストンH3をモノメチル化して発現を正に制御すること、SREK1IP1はがん抑制的な機能を持つことを明らかにした(Akiyama et al. Oncotarget, 2015)。現在、SET7/9低発現株におけるエピジェネティック阻害薬の感受性を検討中である。
4.がん幹細胞の解析と治療開発
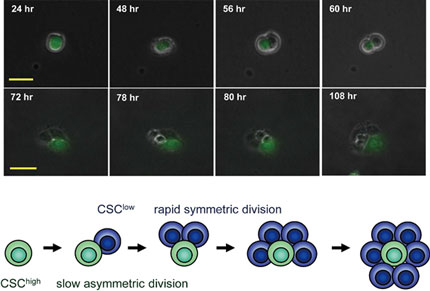
可視化膵がん幹細胞
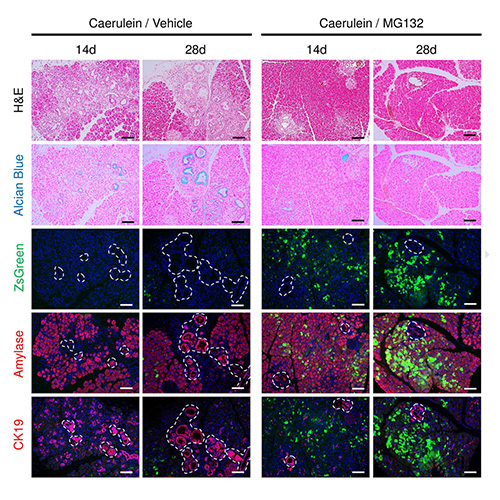
がん幹細胞のプロテアソーム活性非依存性に注目し、蛍光蛋白ZsGreenとプロテアソーム認識モチーフdegronの融合遺伝子(Gdeg)によってがん幹細胞を可視化させ、 その非対称性分裂や抗癌剤耐性の動態を観察することに成功した。可視化ヒト膵がん細胞株を対する化合物スクリーニングによって、がん幹細胞様形質の喪失を誘導する化合物を同定し、 抗癌剤との併用による根治的効果を報告した(Adikrisna, Tanaka et al. Gastroenterology 2012)。 ヒト肝がん細胞の解析では、低酸素下において可視化幹細胞が増加すること、活性酸素を抑制すること、播種転移能を示すことを明らかにした (Muramatsu, Tanaka et al. Hepatology 2013)。 現在、がん幹細胞の転移規定因子とエピゲノム制御メカニズムを解析している(Ito, Tanaka et al. PLoS One 2016)。
プロテアソーム活性可視化マウスモデル
我々は、生体内のプロテアソーム活性を直接観察するために、蛍光蛋白ZsGreenとプロテアソーム認識モチーフdegronの融合遺伝子Gdegをもつ遺伝子改変マウス(Gdegマウス)を作製した(Furuyama, Tanaka et al. Sci Rep 2016)。Gdegマウスの膵臓を観察したところ、正常の膵臓はプロテアソーム活性は低いことが判明した。そこで、GdegマウスにPdx1-Cre;LSL-Kras